Download

1 / 44
700 likes | 2.13k Views
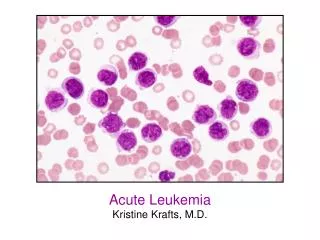
Acute lymphoblastic leukemia/lymphoma (ALL). A group of neoplasms of B or T lymphoblasts Pre-B cell (85%): childhood acute leukemias Pre-T cell: lymphomas in adolescent males, 50~70% with mediastinal (thymic) masses B & T : similar histology, immunophenotype

E N D
Acute lymphoblastic leukemia/lymphoma (ALL) • A group of neoplasms of B or T lymphoblasts • Pre-B cell (85%): childhood acute leukemias • Pre-T cell: lymphomas in adolescent males, 50~70% with mediastinal (thymic) masses • B & T : similar histology, immunophenotype • Chromosome anomaly (90%): hyperdiploidy, pseudodiploid, t(12;21), t(9;22), and t(4;11)
Clinical features of ALL • Blasts in BM suppress hematopoiesis by physical crowding: anemia, neutropenia, thrombocytopenia • Abrupt stormy onset • Bone pain and tenderness • Generalized lymphadenopathy (LAP), splenomegaly, hepatomegaly • CNS manifestations • Treatment (Tx) & prognosis (Px)
Myeloid Neoplasms • This heterogeneous group of neoplasms is an origin in a progenitor cell that normally gives rise to terminally differentiated cells of the myeloid series (erythrocytes, granulocytes, monocytes, and platelets). • Bone marrow (BM) and other MPS • AML: immature myeloid cells in the BM • MDS: ineffective hematopoiesis & cytopenia • MPD: increased production of terminally differentiated myeloid cells
Acute myelogenous leukemia (AML) • Primarily in adult: peak age, 15~39 y/o • Pathophysiology: acquired genetic alteration inhibition of terminal differentiation physical replacement pancytopenia • Tx: clear the BM leukemic clone (cytotoxic drugs), overcome the block in differentiation • Classification • Diagnosis: >20% myeloid blasts in BM, variable number of leukemic cells in PB (aleukemic leukemia)
Chromosomal Abnormalities of AML • 90% AML, with prognostic implication • Ch 5 or 7 deletion: AML after MDS of CT/RT • AML M3 (APL): t(15;17) RAR-α/PML fused gene hybrid mRNA abnormal retinoic acid receptor block myeloid cell differentiation => all-trans-retinoic acid causing neoplastic promyelocytes neutrophils (differentiation therapy), but all patients ultimately relapse
Clinical Features of AML • Weeks or a few months of the onset of symptoms with findings related to pancytopenia, similar to that of ALL • APL: DIC (disseminated intravascular coagulation) • M4 &M5: infiltration of the skin (leukemia cutis) and the gingiva • localized mass composed of myeloblasts: granulocytic sarcoma (myeloblastoma, chloroma)
Myelodysplastic Syndrome (MDS) • A group of clonal stem cell disorders characterized by maturation defects resulting in ineffective hematopoiesis and an increased risk of transformation to AML • BM replaced by the clonal progeny of a mutant multipotent stem cell that retains the capacity to differentiate into RBC, WBC, PLT, but in an ineffective and disordered manner • BM: hyper-/normo-cellular, PB: pancytopenia • Idiopathic (primary) or therapy-related (t-MDS)
MDS • Pathogenesis: unknown (? Stem cell damage) • monosomy 5/7, 5q/7q deletions, trisomy 8, 20q deletion • MF: dysplastic differentiation & <30% blasts –BM: ring sideroblasts, megaloblastoid maturation, nuclear budding; pseudo-Pelger-Huet cells, hypo- or hyper-lobated nucleated megakaryocytes– PB: pseudo-Pelger-Huet cells, giant PLT, macrocyte, poikilocyte, monocytosis
Clinical Course of MDS • Adult >60 y/o • S/S: pancytopenia, asymptomatic (half) • multiple chromosomal abnormalities and severity of cytopenia • median survival: 9~29 months (may >5 yrs) • 10~40%: AML transformation • t-MDS: 4~8 months, more grim prognosis • Tx: allogeneic BMT for younger patients; supportive treatment for the older
Chronic Myeloproliferative Disorders (MPD) • A group of disorders of multipotent progenitor cell capable of terminal differentiation => hypercellular BM and increased hematopoiesis & PB counts, extramedullary hematopoiesis & splenomegaly, later in spent phase (may progress to AML, especially in CML) • CML, PCV (PV), ET, IM • non-specific pathologic findings, overlap with one another and some reactive hyperplasia • Dx: clinical+morphologic+cytogenetic
Chronic Myelogenous Leukemia • Age: 25~60 y/o (peak 30+~40+y/o) • Genetics: Philadelphia chromosome (Ph1)>90% • MF: hypercellular BM with sea-blue histiocytes; leukocytosis, eosinophilia, and basophilia in PB; neoplastic extramedullary hematopoiesis (splenomegaly) • S/S: anemia, hypermetabolism, LUQ distension or pain, lack of leukocyte alkaline phosphatase • Course: slow progression (median survival 3 yrs); accelerated phase (50%) 6-12 m blast crisis 70% AML, 30% ALL (most early B lineage) • Tx: low-dose CT, allogeneic BMT (75% cure)
PCV: polycythemia vera • A multipotent myeloid stem cell neoplasm • absolute increase in red cell mass (Hb, Hct…) • serum erythropoietin: virtually undetectable • BM: hyperplasia fibrosis (spent phase) • Age: 60 y/o; S/S: increased red cell mass hematocrit total blood volume vascular stasis (hypertensive, cyanotic, pruritus, gout…) • risk of bleeding and thrombotic episodes • Px: 10 yrs, spent phase (M with MM), 2~15% to AML; Tx: phlebotomy
ET: essential thrombocytosis • The least common form of MPD • Megakaryocytic hyperplasia: PLT>600K • Dx: by exclusion of other MPDs or reactive • BM: mild to moderate hyperplasia with marked megakaryocytic hyperplasia • PB: giant PLT, mild leukocytosis • S/S: thrombosis and hemorrhage • Px: indolent disorder, long asymptomatic periods, median survival: 12~15 yrs
IM: myelofibrosis with myeloid metaplasia (MMM) • Hallmark: early marrow fibrosis (myelofibrosis) • neoplastic megakaryocytes release PDGF & TGF- fibrosis hematopoietic stem cells seed the spleen, liver, LNs EMH (MM) • BM: early hypercellularity, megakaryocytic hyperplasia and dysplasia, minimal fibrosis hypocellularity, diffuse fibrosis or osteosclerosis • Spleen: huge, EMH with large, clustered megaK • PB: leukoerythroblastosis, dacryocytes
Clinical Course of MMM • Age: 60 y/o or more • S/S: progressive anemia or huge spleen, hyperuricemia and secondary gout • Lab: moderate to severe anemia (normochromic and normocytic) with leukoerythroblastosis, WBC (variable) & PLT (N or elevated, then decreased) • median survival: 1~5yrs, complication: infection, thrombosis, bleeding, 5~20% to AML
Langerhans cell histiocytosis (Histiocytosis X) • Clonal proliferation of the antigen-presenting dendritic cells (normal in the skin and others) • Three categories:- Letterer-Siwe disease: acute disseminated- Hand-Schuller-Christian disease: calvarial defects, diabetes insipidus, and exophthalmos- Eosinophilic granuloma • EM: HX bodies (Birbeck granules)