Download

1 / 49
850 likes | 2.14k Views
Overview of Inherited Metabolic Disorders. Pediatric Resident Academic Half Day. Outline. Overview of genetic / metabolic diseases Overview of cell metabolism Amino acids Glucose homeostasis Fatty acids Complex molecule biosynthesis & degradation Energy metabolism

E N D
Overview of Inherited Metabolic Disorders Pediatric Resident Academic Half Day
Outline • Overview of genetic / metabolic diseases • Overview of cell metabolism • Amino acids • Glucose homeostasis • Fatty acids • Complex molecule biosynthesis & degradation • Energy metabolism • Approaches to treatment • Example case histories for discussion
Genetic disorders of the body’s biochemistry that can cause: Death disability Our goal is: prevention of these outcomes by early diagnoses and treatment Primary, secondary & tertiary prevention How expensive is this? What Are Genetic Metabolic Disorders?
Inborn Errors of Metabolism(Genetic / Metabolic Disorders) Genetic deficiencies in production of proteins: • Enzymes • Transport proteins • Receptor proteins • Sub-cellular organelles: • structural, assembly & chaperone proteins
Overview of Inherited Metabolic Disease • over 700 separate IEM described • most present early: in utero 8 % birth - 1 yr 55 % 1 yr-puberty 32 % adulthood 5 % • for many, early detection prior to irreversible pathology may permit intervention with diet or medical therapy to prevent long-term death or disability • approaches to early detection: • symptomatic presentation • screening • IEM affect about about 1/1000 to 1/2000 persons
Classification by Pathogenic Mechanism • IEM that lead to an acute or progressive intoxication from accumulation of toxic compounds proximal to the metabolic block ( PKU,UCD,MMA,IVA,galactosemia etc.) • IEM with symptomsdue to partial deficiency in energy production ( GSD’s, B-oxidation defects, mitochondrial disorders, congenital lactic acidosis etc.) • IEM that have: • disturbed biosynthesis of complex molecules( CDGS) • disturbed degradation of complex molecules (MPS, GM1 gangliosidosis, Tay-Sach’s/Sandhoff)
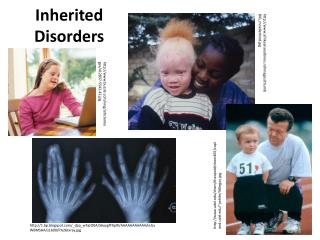
UntreatedPhenylketonuria Hurler-Scheie Syndrome Sandhoff Disease
Overview of Intermediary Metabolism as It Relates to Inherited Metabolic Diseases
Key Metabolic Functions That Our Bodies Must Do: • Accept dietary nutrients and supply them to appropriate body tissues in sufficient but non-toxic amounts • maintain appropriate biosynthetic mechanisms to convert dietary nutrients into required metabolites • maintain metabolic homeostatic mechanisms to ensure that critical nutrients are available as necessary • ensure optimum levels of nutrients by controlling absorption, degradative metabolism and elimination (renal, GI, biliary etc.) • provide mechanisms to support tissue turnover / growth
Genetic Metabolic Disorders Can Cause Disruption of any of these Essential Processes The particular process disrupted determines the clinical outcome in a particular patient Mechanisms of Disruption Include: • toxicity due to excessive metabolite levels (PKU) • inadequate essential precursors (SLOS) • inadequate energy production (mitochondrial disorders) • abnormal biosynthesis of macromolecules (CDGS) • abnormal macromolecule degradation (LSD / peroxisomes) • abnormal transport (cystinuria, cystinosis)
Branched Chain Amino Acid Metabolism:Leucine & Isovaleric Acidemia Isovaleryl-CoA Dehydrogenase Deficiency “Isovaleric Acidemia”
Maintainence of Euglycemia during Fed & Fasting States Maintenance of blood and tissue glucose levels is critical for function • CNS function (except in the infant, CNS is almost completely dependent on glucose from the blood for energy • other tissues also require glucose but can utilize other energy sources as well ie fatty acids and amino acids, glycerol and lactate
Requirements to Maintain Euglycemia Under “Fasting” Conditions • Functioning hepatic gluconeogenic & glycogenolytic enzyme systems • adequate endogenous gluconeogenic substrates (amino acids, glycerol, lactate) • adequate B-oxidation of fatty acids to synthesize glucose & ketones • functional endocrine system to modulate & integrate the above system components
FED STATE High GI absorption High Glycogen biosynthesis High triglyceride biosynthesis Low gluconeogenesis Low lipolysis FASTING STATE Low GI absorption High Glycogenolysis High Lipolysiswith mobilization of fatty acids & ketones High Gluconeogenesis Homeostatic Processes Maintaining Euglycemia(insulin & glucagon in response to glucose levels)
Phases of Glucose Homeostasis 1.Glucose absorptive phase: 3 - 4 hrs after glucose ingestion (high insulin) 2.Post absorptive/early starvation: 3-12 hrs glucose (from hepatic glycogen) to brain, RBC, renal medulla 3. Early / Intermediate Starvation: 14+ hrs gluconeogenesis & (later) lipolysis
GSD-II ( lysosomal) GSD-IV GSD-V,GSD-VI,GSD-IX GSD-0 GSD-III GSD-1a&b GSD-VII GSD-X,GSD-XII,GSD-XIII GSD-XI (LDH) LIVER MUSCLE
VLCAD,MCAD, SCAD Trifunctional protein
Biosynthesis & Degradation of Complex Molecules Considerable energy and substrates are used in cells for the synthesis and degradation of macromolecules that: • Perform biological functions • Become components of sub-cellular structures
Endoplasmic Reticulum: Synthesis of GlycoproteinsN-Glycosylation & the Mannose Pathway
Abnormal glycopeptide BiosynthesisDisorders of N-Glycosylation
Abnormal Glycopeptide BiosynthesisO-Glycosylation and its Disorders
Metabolic Role of Lysosomes • Degradation of endogenous and exogenous macromolecules • Acidic hydrolysis: • Molecules include: mucopolysaccharides sphingolipids peptides oligosaccharides glycopeptides lipids S-acetylated proteins monosaccharides/aminoacids/monomers
Typical Lysosomal Storage Disease History • Initially “clinically normal” • Slow onset of symptoms usually involving multiple organs / systems • Progressive deterioration • Usually premature death • Typical features often include: neurodegeneration, organ enlargement, connective tissue involvement, cardiac & pulmonary involvement, other organs (vascular endothelium, muscle, kidney)
Sphingolipidoses: Tay-Sach’s, Sandhoff, GM1 gangliosidosis, MLD,Krabbes, Fabry, Gaucher, Farber, Niemann-Pick Mucopolysaccharidoses: Hurler/ Hurler-Scheie/Scheie, Hunter, San Filippo, Morquio, Maroteau-Lamy, Sly Glycogenoses Pompe disease Lipid Storage diseases Wolman, cholesterol ester, NP”C” Oligosaccharide/glycopeptidoses Mannosidoses, fucosidosis, Schindlers, sialidoses, aspartylglycosaminuria Multiple enzyme deficiencies I-cell & MLIII, multiple sulfatase deficiency, galactosialidosis Transport deficiencies Cystinosis, Salla disease, ISS Peptide Storage Diseases Pycnodysostoses, infantile NClF 40+ Lysosomal Storage Diseases Identified
Metabolic Jobs of Mitochondria • Amino acid metabolism • Urea cycle ( removal of ammonia) • Steroid biosynthesis • Fatty acid oxidation ( carnitine, B-oxidation) • Ketone body metabolism • Carbohydrate metabolism (PDH) • Aerobic energy product’n
Mitochondria: Electron Transport Chain Enzyme ComplexesATP produced in using the respiratory chain Respiratory chain (inner compartment) (Five multimeric complexes + two electron carriers) • Complex I: 46 subunits ( 7 mDNA + 39 nDNA) • Complex II: 4 subunits ( 4 nDNA) • Coenzyme Q10 (ubiquinone) - carrier to complex III) • Complex III: ( 11 subunits (1 mDNA – 10nDNA) • Cytochrome C - mobile carrier to complex IV • Complex IV: 13 subunits (3 mDNA – 10 nDNA) Protons extruded by Cplx’s I,II, III, & IV • Complex V: ATP synthase – “Couples” proton reintake which is coupled to ATP synthesis
TCA Cycle & Respiratory Chain Glucose TCA Cycle Electron Transport Chain
Energy Production in Mitochondria Glycolysis, pyruvate, aconitate, Malate + other dehydrogen’n Rx’s NAD-H2 Cplx I H+ NAD (CoQ10) FAD-H2 H+ Cplx II Succinate, Isol, Val, Met, Thr, SCFA’s (CoQ10) FAD Cplx III ETF / ETF-QO (Cyt-C) H+ Cplx IV Fatty .Acid B-oxid’n, dimethylglycine, sarcosine O2 H20 ADP H+ Cplx V ATP Inner Mitoch. Membrane
Mitochondria • Only organelle other then nucleus that has: • DNA (circular / double stranded) - 16,569 bases • Can synthesize own RNA & proteins • mDNA– 37 genes • 24 for translation (2 rRNA / 22 tRNA) • 13 for proteins of Respiratory Chain subunits • nDNA – many genes • code for 1000+ mitochondrial proteins (structural, transport, chaparone & enzyme)
Any significant defect can lead to deficient function and result in clinical abnormality • Based on physiological function(s) affected • Based on organ(s) affected • Based on severity of mutation and resulting deficiency of protein-mediated biochemical function • Recognition often difficult clinically and usually requires laboratory support for screeening, diagnosis and treament.
Common Treatment Examples • Restriction / supplements / medications • PKU& other aminoacidopathies • Urea cycle disorders • Organic acidopathies (MMA,PA, IVA etc.) • Ensure nutrient availability • Glycogen storage disorders • B-oxidation disorders • Enhancement of organelle function • mitochondrial disorders • Cell / organ replacement • lysosomal storage disorders
More Recent Approaches to Therapy • End organ protection: large chain neutal amino acids in PKU • Stabilization of “mis-folded” proteins: otherwise that would be recognized as having defective “folding” and removed via proteosome mechanism • Improved correction of biochemical milieu in cells of patient with the metabolic defect:
End Organ Protection in PKU CNS CNS High PHE Lower PHE BBB BBB Isol Leu Val Tyr Trypt Met High plasma phenylalanine High plasma phenylalanine PreKunil Low PHE Diet
Indirect Therapy: Replacement of Essential Metabolites PKU: Extra tyrosine for protein synthesis, neurotransmitter biosynthesis, pigment biosynthesis Urea Cycle Disorders: Extra arginine to maintain adequate levels of urea cycle intermediates “ Many IEM Diets require Further Modification” Urea Cycle Disorders May need increased leucine, isoleucine & valine to compensate for loss of “N” as phenylacetyl-glutamine
Organ Transplantation(to provide metabolic capability) Liver • Familial Hypercholesterolemia (LDL-cholesterol receptor deficiency) • Tyrosinemia • Glycogen Storage Disease (Type I) • Primary hyperoxaluria * Kidney • Fabry Disease • Cystinosis • Primary hyperoxaluria * Bone Marrow • Various lysosomal storage diseases ie. Hurler syndrome (MPSI) Cornea • Cystinosis, Fabry disease
Biopterin-responsive PKU(PAH Deficiency) • Not due to a biopterin biosynthesis disorder • Up to 1/3 of PKU patients (usually milder variants) • Will have higher tolerance for PHE in diet when on BH4 OR • Be able to avoid low-PHE diet • Clinical trials now in process
Lysosomal Storage Disorders:Treatment options • Supportive care • Enzyme replacement therapy • Substrate depletion (biosynthesis inhibitors) • Hematopoeitic stem cell transplant • Chaperone Therapy (research only) • End organ protection therapy (research only) • Gene therapy
Enzyme Replacement Therapy vrs. Substate Biosynthesis Inhibition Biosynthesis Degradation LYSOSOME Glucosylceramide Glucosylceramide Biosynthesis Inhibitor ERT Cellular Damage
“Chaperone” Therapy Protein Biosynthesis in RER Endoplasmic Endoplasmic protein modification & folding Misfolded Properly folded Degradation via Ubiquitin plus proteosome system Transport from trans-GOLGI to lysosme with activation at acidic pH
Case Histories • Case 1 – Positive Newborn Metabolic Screen • Case 2 – Hepatomegaly with abnormal liver pathology • Case 3 – 18 month boy with hepatomegaly and obtundation • Case 4 – 5 year girl with hearing loss & macrocephaly • Case 5 – 10 month boy with developmental delay & dysmorphic facies