Download

1 / 43
530 likes | 1.29k Views
Amino acid metabolism 1. Dr.S.Chakravarty,MD. Learning objectives . Explain the steps in synthesis of various non-essential amino acids in the body List the molecules derived from aromatic amino acids and their uses

E N D
Amino acid metabolism 1 Dr.S.Chakravarty,MD
Learning objectives • Explain the steps in synthesis of various non-essential amino acids in the body • List the molecules derived from aromatic amino acids and their uses • Discuss the enzyme deficiencies of aromatic amino acid metabolism and their clinical features • Differentiate various types of phenylketonuria and its diagnosis • Discuss the clinical features of Alkaptonuria and its treatment
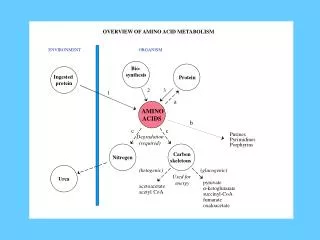
Biosynthesis of Non Essential Amino-acids
1. Glutamate : NH3 Glutamate dehydrogenase Alpha ketoGlutarate Glutamate Glutamate Dehydrogenase 2. Glutamine: NH3 Glutamate Glutamine Glutamine syhthetase Glutamine synthase
Transamination reactions 3. Alanine: ALT Pyruvate Alanine PLP Glutamate Alpha ketoglutarate 4. Aspartate : AST Oxaloacetate Aspartate PLP Glutamate Alpha ketoglutarate
5. Asparagine: NH3 Aspartate Asparagine Asparaginesynthase 6. Tyrosine : THB DHB Tyrosine Phenylalanine Phenyl alanine Hydroxylase
7. Glycine- Glycineamidotransferases synthesize glycine from glyoxylate and glutamate or alanine. Two other reactions make glycine :- serine hydroxymethytransferase reaction(freely reversible)
8. Serine :- Two ways to make it • Reversal of serine hydroxymethytransferase reaction
9. Proline – from Glutamate -reversal reaction of proline catabolism
Methionine THF Methionineadenosyltransferase ATP 10. Cysteine: S- AdenosylMethionine Acceptor CH3-acceptor Methyl transferase Homocysteine methyl Transferase S- AdenosylHomocystiene (CH3 1-carbon )B12 Homocysteine Serine Methyl THF B6 Cystathionineβsynthase Cystathionine B6 Cystathioninase Cysteine Alpha-keto butyrate
Metabolism of Aromatic amino acids
Metabolism of Aromatic amino acids: • Phenylalanine – essential • Tyrosine – non essential • Tryptophan – essential
PHENYLALANINE AND TYROSINE
FATES OF TYROSINE IN BODY MELANIN SYNTHESIS SYNTHESIS OF THYROID HORMONES TYROSINE CATABOLISM TO ACETOACETIC ACID (KETOGENIC ) + FUMARIC ACID SYNTHESIS OF CATECHOLAMINES eg Epinephrine and Norepinephrine
SYNTHESIS OF TYROSINE FROM PHENYLALANINE Dihydrobiopterinreductase Phenylalanine hydroxylase reaction
Catecholamine synthesis Phenylalanine Phenylalanine hydroxylase Tyrosine THB Dihydrobiopterinreductase Tyrosine Hydroxylase DHB DOPA Important – alpha methyl DOPA inhibits dopadecarboxylase and prevents hypertension by decreasing epinephrine co2 DOPA Decarboxylase B6 Dopamine ArvidCarlsson M.D. Nobel Prize 2000 alongwith Eric Kandel and Paul Greengard. Ascorbate(Vitamin C) O2, Cu2+ Dopamine βoxidase Dehydro-Ascorbate Nor-Epinephrine S- AdenosylMethionine (SAM) Phenylethanolamine N-methyl Transferase (NMT) S- AdenosylHomocysteine (SAH) Epinephrine Catechol-o-methyl Transferase (COMT) mono amine oxidase(MAO) Vanillylmandelic acid metanephrine CNS and ADRENAL MEDULLA
Diseases associated with catecholamine synthesis: • Schizophrenia – Dopamine overproduction • Parkinson’s disease : Damage to Nigro-striatal tract - Dopamine Treatment: – Levo-DOPA + Carbidopa Carbidopa is PERIPHERAL DOPA-DECARBOXYLASE INHIBITOR it increases the plasma half-life of levodopa from 50 minutes to 1½ hours. Carbidopa cannot cross the blood brain barrier, so it inhibits only peripheral DDC. It thus prevents the conversion of L-DOPA to dopamine peripherally • Pheochromocytoma • Neuroblastoma Increased catecholamine production
IMPORTANCE OF VMA estimation • Some tumors like Pheochromocytoma (epinephrine excess ) or Neuroblastoma • Excess of VMA in urine Lab analysis
Formation of Melanin Tyrosine Copper Tyrosinase (Melanoblasts ) TYROSINASE IS ABSENT IN ALBINISM NO MELANIN DOPA Tyrosinase (Melanoblasts ) Copper Dopaquinone Several steps Melanin THIS ALSO EXPLAINS HYPOPIGMENTATION IN PHENYLKETONURIA !!
Formation of thyroid hormones • T3Triiodothyronine • T4Thyroxine
Catabolism of phenylalanine and tyrosine USMLE !! Phenylalanine Phenylketonuria Phenyl alanine Hydroxylase Tyrosine Tyrosinemia- II Tyrosine Aminotransferase PLP Parahydroxyphenylpyruvate P-Hydroxyphenylpyruvate hydroxylase Cu, Vit C Homogentisic acid Alkaptonuria Homgentisateoxidase Tyrosinemia- I Fumarylacetoacetatehydrolase Fumarate Acetoacetate
CATABOLISM OF TYROSINE 1 Type II TYROSINEMIA 2 NEONATAL TYROSINEMIA ALKAPTONURIA 3 TYPE I TYROSINEMIA 4
Tyrosinemia Type 1 • Defect in fumarylacetoacetatehydrolase • Plasma tyrosine levels elevated (6-12mg/dl)] • ACUTE FORM – FATAL BY 6-8 MONTHS • CHRONIC FORM- 10 YEARS • DIARRHOEA • VOMITING • CABBAGE LIKE ODOR • LIVER FAILURE • URINE :-tyrosine, p -hydroxyphenylpyruvate, P-hydroxyphenyl lactate , p-hydroxyphenyl acetate
Tyrosinemia Type II(RichnerHanhart Syndrome) DEF. OF TYROSINE AMINOTRANSFERASE • Mental retardation • Keratosis of palmar surface • Painful corneal lesions • Photophobia NEONATAL TYROSINEMIA • Def . Of p-hydroxyphenylpyruvate hydroxylase
Phenylketonuria Phenylalanine Phenylalanine hydroxylase defect (-) Transaminase Dihydrobiopterinreductase defect Tyrosine Phenylpyruvate Reduction ALTERNATE ROUTES OF METABOLISM OF PHENYLALANINE Decarboxylation Phenyl Lactate Phenyl acetate Conjugation with glutamine Phenyl acetyl Glutamine
USMLE !! Phenylketonuria • Autosomal recessive disease – MC disorder of amino acid metabolism • Def of phenylalanine hydroxylase or Dihydrobiopterinreductase. • Increased phenylalanine in the blood • Saturates – LNAAT (large neutral aminoacid transporter system of brain). • mental retardation, seizures- • Poor protein and neurotransmitter synthesis in brain • Toxicity from accumulating alternate metabolites like phenylketones • Decreased pigmentation of skin and eyes.
Amino Acid Disorders Argininosuccinicaciduria (ASA) Citrullinemia, type I (CIT) Classic phenylketonuria (PKU) Homocystinuria (HCY) Maple syrup urine disease (MSUD) Tyrosinemia, type I (TYR I) Tyrosinemia, type II (TYR II) Endocrine Disorders Congenital adrenal hyperplasia (CAH) Primary congenital hypothyroidism (CH) Fatty Acid Oxidation Disorders Carnitineacylcarnitinetranslocase deficiency (CACT) Carnitinepalmitoyltransferase I deficiency (CPT-IA) Carnitinepalmitoyltransferase type II deficiency (CPT-II) Carnitine uptake defect (CUD) Glutaricacidemia, type II (GA-2) Long-chain L-3 hydroxyacyl-CoAdehydrogenase deficiency (LCHAD) Medium-chain acyl-CoAdehydrogenase deficiency (MCAD) Short-chain acyl-CoAdehydrogenase deficiency (SCAD) Trifunctional protein deficiency (TFP) Very long-chain acyl-CoAdehydrogenase deficiency (VLCAD) NEONATAL SCREENING IN FLORIDA (SOURCE CDC website and http://www.babysfirsttest.org) Hemoglobin Disorders S, Beta-thalassemia (Hb S/ßTh) S, C disease (Hb S/C) Sickle cell anemia (Hb SS) Organic Acid Conditions 3-Hydroxy-3-methylglutaric aciduria (HMG) 3-Methylcrotonyl-CoA carboxylase deficiency (3-MCC) Beta-ketothiolase deficiency (BKT) Glutaricacidemia type I (GA1) Holocarboxylasesynthetase deficiency (MCD) Isovalericacidemia (IVA) Methylmalonicacidemia (cobalamin disorders) (Cbl A,B) Methylmalonicacidemia (methymalonyl-CoAmutase deficiency) (MUT) Propionicacidemia (PROP) Other Disorders Biotinidase deficiency (BIOT) Classic galactosemia (GALT) Cystic fibrosis (CF) Hearing loss (HEAR) Severe combined immunodeficiency (SCID) Cont.. • Mousy/ Musty odor of urine – phenyl acetate, phenyl lactate and phenylpyruvate in urine. • National biochemical screening programme • Blood sample – Heel filter paper analytical laboratory ( PCR + HPLC or TANDEM MASS SPECTROMETRY) • Screens diseases like :- • Cystic fibrosis • PKU • Congenital hypothyroidism • Medium chain acylCoAdehydrogenase deficiency • FeCl3 test – Ferric chloride test. • Guthrie test: Gold standard of the past • Certain strains of Bacillus Subtilis need Phe as essential growth factor.Bacterial growth cannot occur in medium devoid of Phe. • So, bactera will grow if blood containing Phe is added = PHENYLKETONURIA
Treatment • Early detection is VERY IMPORTANT !! • Diet containing low phenylalanine • ( but NEVER ZERO Phe!!) • FOOD BASED ON TAPIOCA (CASSAVA ) IS HELPFUL • SPECIAL DIET TILL 5YEARS OF AGE • SPECIAL DIET AGAIN IF PERSON IS PREGNANT LATER ON Excess Phe affects brain development of fetus .
Phenylketonuria Phenylalanine Hydroxylase def Dihydrobiopterinreductase def Low levels of dopamine High levels of prolactin Low levels of cathecolamines Increased tryptophan and decreased serotonin • Normal levels of dopamine • Normal levels of prolactin • Normal levels of catecholamines • Normal levels of tryptophan and serotonin
Alkaptonuria • Autosomal Recessive • Deficient enzyme: Homogentisate 1,2-dioxygenase/ (Oxidase) • conversion of homogentisic acid (product of tyrosine metabolism) to maleylacetoacetate (→ acetoacetate → Fumarate → TCA) • Pathology • Homogentisic acid accumulates, auto-oxidizes • Oxidized homogentisate polymerizes, forms dark-colored pigment • Purplish black color of urine on standing • Precipitates of dark homogentisic acid (Alkaptan bodies) deposit in connective tissue discoloration (ochronosis) • e.g. in cartilage, joints, ear wax • vertebrae • deposits cause Arthralgia (joint pain) • sometimes associated with degenerative arthritis
Main Fates of Tryptophan Tryptophan I N D I C A N Niacin Catabolized to AcetoacetylCoA (KETOGENIC ) + alanine (Glucogenic ) Synthesis of Serotonin and Melatonin
Tryptophan THB NADP Tryptophan Hydroxylase DHB NADPH + H + 5-HydroxyTryptophan PLP A.A. decarboxylase Monoaminooxidase-A (MAO) 5-HydroxyTryptamine (Serotonin) Acetyl CoA 5 HYDROXY INDOLE ACETIC ACID ( HIAA) Acetylation MELATONIN SYNTHESIS Acetylation N-acetyl Serotonin SAM methylation SAH Melatonin
Catabolism of serotonin Serotonin MAO-A inhibitors (Anti-Depressants) Monoaminooxidase-A (MAO) (-) 5- Hydroxyindole acetic acid (HIAA)
MAO- mono amino oxidase • Epinephrine, norepinephrine, serotonin and melatonin are metabolised by MAO- A enzymes • Dopamine, Tyramine and tryptamine are metabolised by both MAO-A and MAO-B • Tyramine mimics catecholamines in their actions
Cheese reaction • A patient presents with headaches, palpitations, nausea and vomiting and elevated blood pressure. These symptoms appear after the person has eaten a large meal containing aged cheeses and wine. The patient’s history indicates that he is on some medicaton for a different condition. Assuming that the medication is in some way involved in these symptoms, which enzyme might be the target of this drug? • Glutamate decarboxylase • Monoamine oxidase • Tyrosine hydroxylase • DOPA decarboxylase • COMT (catechol O-methyl transferase)
Tryptophan Tryptophan catabolism Tryptophan pyrrolase N-formylkynurenine THFA Kynurenineformylase Formyl THFA 1 CARBON POOL 3-hydroxykynurenine Xanthurenicacid H2O Kynureninase PLP Alanine 3-hydroxy Anthranilic acid(HIAA ) TCA cycle Acetyl Co-A NAD, NADP (Niacin) Acetoacetyl Co-A 60mg tryptophan = 1 mg NIACIN
Diseases associated with tryptophan Metabolism • Carcinoid syndrome:- (Argentaffinomas ) • Neuroendocrine tumors – Midgut, bronchus • Excessive serotonin and kallikrenin. • Diarrhoea , flushing, abdominal cramps, • Heart failure – damage to valves • Diagnosis : HIAA in urine
Pellagra like synptoms: Def of B6 or Tryptophan Diarrhoea, Dementia and Dermatitis • Remember – Hartnups disease • Melatonin: promotes sleep – sleep wake cycle • Hormone of the dark – blue light inhibits melatonin synthesis. • Lowers Leptin levels • Tryptophan load test – B6 deficiency
Depression • Decrease in serotonin levels in CNS • Treatment : • MAO-A inhibitors • SSRIs – selective serotonin reuptake inhibitors
Mcq • The non essential amino acid that becomes essential in PKU is :- • A. Phenylanaline • B.Tyrosine • C.Tryptophan • D. ALANINE • E. Cysteine
Mcq • The cause of light skin color in PKU is • A. decreased synthesis of melanin from Phe • B. decreased synthesis of melanin from Tyr • C. excess melanin synthesis from Phe • D. excess of phenylketones • E.mental retardation causes decreased melatonin