Download

1 / 37
390 likes | 728 Views
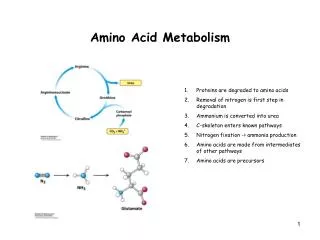
Amino Acid Metabolism (day-2). What to Know. What is the Metabolic Fate of Ammonium? How is Escherichia coli Glutamine Synthetase regulated? Understand general ways that organisms synthesize amino acids Know the definition of essential versus nonessential amino acids

E N D
What to Know • What is the Metabolic Fate of Ammonium? • How is Escherichia coli Glutamine Synthetase regulated? • Understand general ways that organisms synthesize amino acids • Know the definition of essential versus nonessential amino acids • Understand the glutamate transaminase rxn • Have a general understanding of the 3 key metabolic pathways responsible for amino acid synthesis • Understand the key features of amino acid degradation with regard to the citric acid and urea pathways • General understanding of classroom discussion of PKU, albinism, parkinson’s and porphyrias
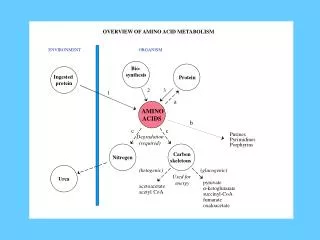
Fate of Ammonium Ions • Only three major reactions introduce NH4 into cells: • Glutamate dehydrogenase • Glutamine synthetase • Carbamoyl-phosphate synthetase I (mitochondrial enzyme of urea cycle)
Glutamine Synthetase (a) The glutamine synthetase reaction. (b) The reaction proceeds by activation of the γ-carboxyl group of Glu by ATP, followed by amidation by NH4+.
Carbamoyl-Phosphate Synthetase • Reaction is: NH4+ + HCO3- + 2ATP → H2N-COO-PO32- + 2ADP + Pi + 2H+ • This reaction is an early step in the urea cycle • Note the name “synthetase”, which is reserved for synthetic enzymes that use ATP • Enzymes that synthesize but do not use ATP are termed “synthases”
What Regulatory Mechanisms Act on Escherichia coli Glutamine Synthetase? • A Case Study in Regulation • GS (a 600kD α12-dodecahexameric protein) in E. coli is regulated in three ways: • Feedback inhibition • Covalent modification (interconverts between inactive and active forms) • Regulation of gene expression and protein synthesis control the amount of GS in cells • But no such regulation occurs in eukaryotic versions of GS
The subunit organization of bacterial glutamine synthetase. (a) Schematic (b) Molecular structure: note the pairs of metal ions (dark blue) that define the active sites.
Glutamine Synthetase is Regulated by Covalent Modification Covalent modification of GS: Adenylylation of Tyr-397 in the glutamine synthetase polypeptide via an ATP-dependent reaction by the converted enzyme adenylyl transferase.
The cyclic cascade system regulating the covalent modification of GS. Degree of adenylation, n, ~ GS activity so high [Gln] / [α-KG] ratio = cell nitrogen sufficiency and GS becomes adenylated and inactivated.
How Do Organisms Synthesize Amino Acids? • Plants and microorganisms can make all 20 amino acids and all other needed N metabolites • In these organisms, glutamate is the source of N, via transamination (aminotransferase) reactions of α-keto acid analogue of the amino acid • Mammals can make only 10 of the 20 amino acids • The others are classed as "essential" amino acids and must be obtained in the diet • All amino acids are grouped into families according to the intermediates that they are made from – • i.e. Glu, Gln, Pro, Arg are all members of the α-ketoglutarate family because they are derived from citric acid cycle intermediate α-ketoglutarate
Amino Acids Are Synthesized From a Limited Number of Precursors
Major Amino Acid Pathways • Carbon skeletons of all 20 amino acids are derived from just seven metabolic intermediates • The seven intermediates are found in three metabolic pathways: 1) 3 glycolytic pathway intermediates • 2 pentose phosphate pathway intermediates • 2 citrate cycle intermediates Note: Plants and bacteria are capable of synthesizing all 20 of the amino acids illustrated in the figure; yet amino acid biosynthesis in animals is much more restricted due to the lack of many of the required enzymes.
Feedback inhibition plays a pivotal role modulating amino acid biosynthetic pathways
Amino Groups for Amino Acids Are Derived From Glutamate in Transamination Rxns • Amino Acid1 + α-Keto acid2α-Keto acid1 + Amino acid2 Glutamate-dependent transamination of α-keto acid carbon skeletons is a primary mechanism for amino acid synthesis.
Amino Groups for Amino Acids Are Derived From Glutamate in Transamination Rxns Glutamate-dependent transamination of α-keto acid carbon skeletons is a primary mechanism for amino acid synthesis. The transamination of oxaloacetate by glutamate to yield aspartate and α-ketoglutarate is a prime example.
The Aspartate Family of Amino Acids Includes Asp, Asn, Lys, Met, Thr, Ile Aspartate biosynthesis via transamination of oxaloacetate by glutamate.
The Aspartate Family of Amino Acids Includes Asp, Asn, Lys, Met, Thr, Ile Asparagine biosynthesis from Asp, Gln, and ATP by asparagine synthetase. β-Aspartyladenylate is an enzyme-bond intermediate.
Biosynthesis of three nonessential amino acids (alanine, aspartate and asparagine) and six essential amino acids (methionine, threonine, lysine, isoleucine, valine and isoleucine) in E. coli involves two interconnected pathways utilizing pyruvate and oxaloacetate as precursors.
Metabolic degradation of the common amino acids Glucogenic amino acids are shown in pink, ketogenic in blue
Shikimate Pathway Aromatic amino acids are synthesized in plants, fungi and bacteria by a pathway involving formation of a hydrocarbon rain following the condensation of phosphoenolpyruvate and erythrose-4-phosphate. Chorismate is a precursor to the three aromatic amino acids, tryptophan, tyrosine and phenylalanine. Herbicides target chorismate “Roundup” “Roundup Ready soybeans” via gene gun insertion of DNA
Amino Acids are Precursors to other Biomolecules • In 1945, Dr. David Sherman discovered that glycine contributed all for nitrogen’s to team with carbon atoms coming from both glycine and acetate.
Tyrosine Precursor to several important molecules in metabolic signaling and neurotransmission including epinephrine and dopamine
Tyrosine is also the precursor to pigment molecules called melanins that are produced from dopaquinone
Inborn Errors of Metabolism • PKU occurs one in every 15,000 births • Caused by the accumulation of phenylalanine in the blood (30-50 times higher than normal) • The high phenylalanine level leads to production of metabolites such as phenyl pyruvate, phenyl acetate and phenyllactate, all of which are associated with neurological and developmental problems
Phenylketonuriacs must be careful to avoid processed foods and beverages containing aspartame (aspartyl-phenylalanine methyl ester) James Schlatter Phenylalanine hydroxylase gene located on chromosome 12– autosomal recessive genetic disease Probability that two PKU carriers will have a child with the disease is 25% frequency of carriers equals 2%, therefore, probability of a baby born with PKU by random chance equals one in 10,000
Albinism is another autosomal recessive disease Defective gene = tyrosinase tyrosinase deficiency results in loss of hair and skin pigments
Porphyrias • Porphyrias are caused by either autosomal recessive or autosomal dominant mutations. • Inhibit porphyrin ring synthesis
Acute Intermittent Porphyria • Defective gene = porphobilinogendeaminase with dominant genetic phenotype • Under normal conditions disease is often asymptomatic but a variety of factors including hormones, drugs or dietary changes can trigger stomach pain and neurological problems • More rare form = congenital erythropoieticporphyria which causes defective gene encoding uroporphyrinogen-III cosynthase • Leads to several defective metabolites including uroporphyrinogen-1which builds up in the teeth causing them to turn reddish brown and fluoresce under UV light The combination of symptoms of congenital erythropoieticporphyria, combined with common medieval practice of drinking animal blood as a treatment for human ailments may have accounted for the legend of vampires.