Download
1 / 1
10 likes | 168 Views
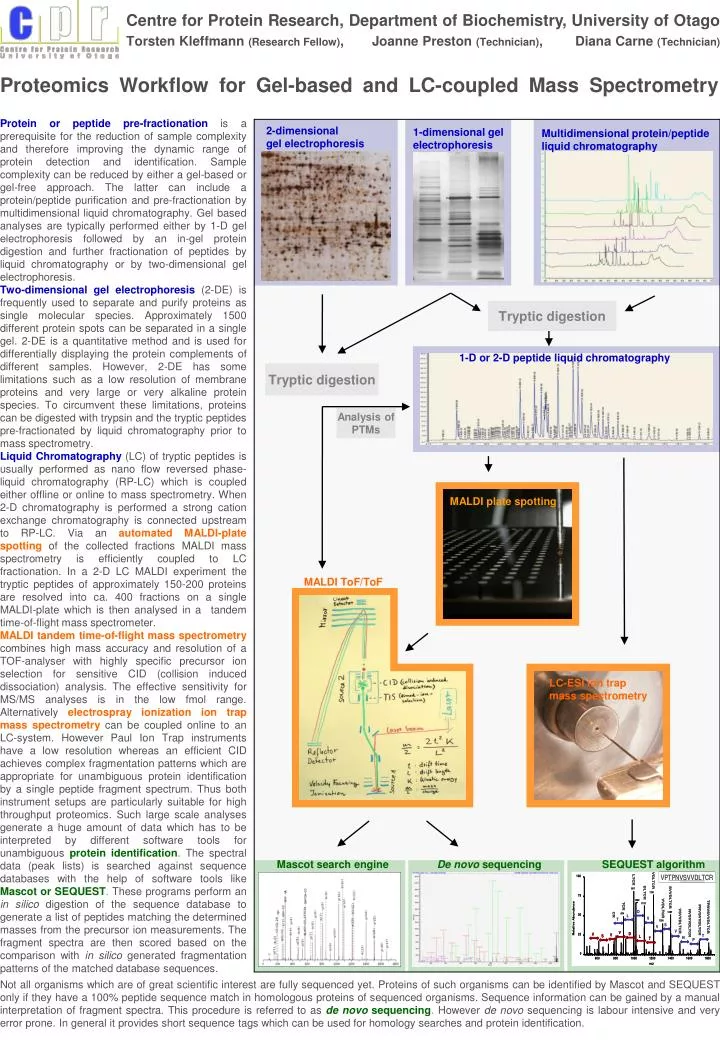
Centre for Protein Research, Department of Biochemistry, University of Otago Torsten Kleffmann (Research Fellow) , Joanne Preston (Technician) , Diana Carne (Technician). Proteomics Workflow for Gel-based and LC-coupled Mass Spectrometry.

E N D
Centre for Protein Research, Department of Biochemistry, University of OtagoTorsten Kleffmann (Research Fellow), Joanne Preston (Technician), Diana Carne (Technician) Proteomics Workflow for Gel-based and LC-coupled Mass Spectrometry Protein or peptide pre-fractionationis a prerequisite for the reduction of sample complexity and therefore improving the dynamic range of protein detection and identification. Sample complexity can be reduced by either a gel-based or gel-free approach. The latter can include a protein/peptide purification and pre-fractionation by multidimensional liquid chromatography. Gel based analyses are typically performed either by 1-D gel electrophoresis followed by an in-gel protein digestion and further fractionation of peptides by liquid chromatography or by two-dimensional gel electrophoresis. Two-dimensional gel electrophoresis(2-DE) is frequently used to separate and purify proteins as single molecular species. Approximately 1500 different protein spots can be separated in a single gel. 2-DE is a quantitative method and is used for differentially displaying the protein complements of different samples. However, 2-DE has some limitations such as a low resolution of membrane proteins and very large or very alkaline protein species. To circumvent these limitations, proteins can be digested with trypsin and the tryptic peptides pre-fractionated by liquid chromatography prior to mass spectrometry. Liquid Chromatography(LC) of tryptic peptides is usually performed as nano flow reversed phase- liquid chromatography (RP-LC) which is coupled either offline or online to mass spectrometry. When 2-D chromatography is performed a strong cation exchange chromatography is connected upstream to RP-LC. Via anautomated MALDI-plate spottingof the collected fractions MALDI mass spectrometry is efficiently coupled to LC fractionation. In a 2-D LC MALDI experiment the tryptic peptides of approximately 150-200 proteins are resolved into ca. 400 fractions on a single MALDI-plate which is then analysed in a tandem time-of-flight mass spectrometer. MALDI tandem time-of-flight mass spectrometrycombines high mass accuracy and resolution of a TOF-analyser with highly specific precursor ion selection for sensitive CID (collision induced dissociation) analysis. The effective sensitivity for MS/MS analyses is in the low fmol range. Alternativelyelectrospray ionization ion trap mass spectrometrycan be coupled online to an LC-system. However Paul Ion Trap instruments have a low resolution whereas an efficient CID achieves complex fragmentation patterns which are appropriate for unambiguous protein identification by a single peptide fragment spectrum. Thus both instrument setups are particularly suitable for high throughput proteomics. Such large scale analyses generate a huge amount of data which has to be interpreted by different software tools for unambiguous protein identification. The spectral data (peak lists) is searched against sequence databases with the help of software tools likeMascot or SEQUEST. These programs perform an in silico digestion of the sequence database to generate a list of peptides matching the determined masses from the precursor ion measurements. The fragment spectra are then scored based on the comparison with in silico generated fragmentation patterns of the matched database sequences. 2-dimensionalgel electrophoresis 1-dimensional gelelectrophoresis Multidimensional protein/peptideliquid chromatography Tryptic digestion 1-D or 2-D peptide liquid chromatography Tryptic digestion Analysis of PTMs MALDI plate spotting MALDI ToF/ToF LC-ESI ion trapmass spectrometry Mascot search engine De novo sequencing SEQUEST algorithm Not all organisms which are of great scientific interest are fully sequenced yet. Proteins of such organisms can be identified by Mascot and SEQUEST only if they have a 100% peptide sequence match in homologous proteins of sequenced organisms. Sequence information can be gained by a manual interpretation of fragment spectra. This procedure is referred to asde novo sequencing. However de novo sequencing is labour intensive and very error prone. In general it provides short sequence tags which can be used for homology searches and protein identification.