Download
1 / 61
610 likes | 827 Views
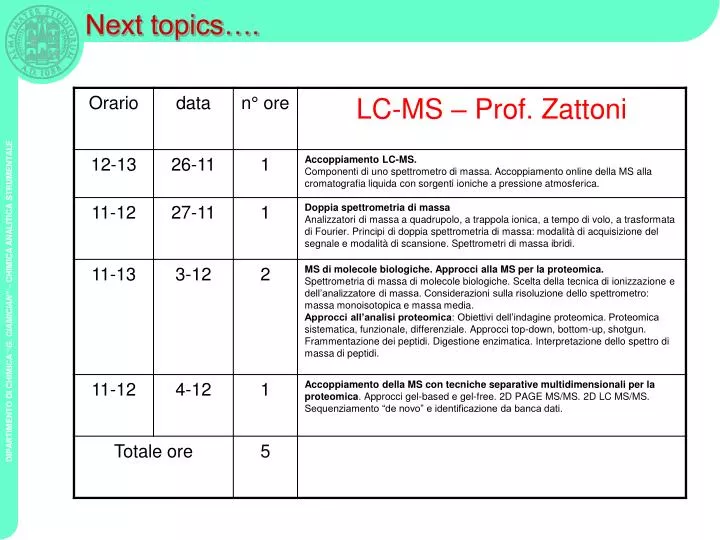
Next topics…. Schema di uno spettrometro di massa. Computer. Segnale. Sistema di introduzione. Sorgente di ioni. Analizzatore di massa. Rivelatore. Campione. Ioni. Ioni. 10 -5 -10 -8 torr. Campione. Sistema di vuoto. Classificazione delle sorgenti ioniche.

E N D
Schema di uno spettrometro di massa Computer Segnale Sistema di introduzione Sorgente di ioni Analizzatore di massa Rivelatore Campione Ioni Ioni 10-5-10-8 torr Campione Sistema di vuoto
Classificazione delle sorgenti ioniche • Sorgenti “dure” (elevata frammentazione) • Ionizzazione elettronica • Sorgenti “molli” (bassa frammentazione) • Ionizzazione chimica • Desorbimento (MALDI, elettrospray) • Fotoionizzazione
Sorgenti ioniche Stato del campione Sorgente Liquido Elettrospray – nanospray APCI (atmospheric pressure chemical ionization) APPI (atmospheric pressure photoionization) LC: flussi ~ 0.1 – 1 cm3 min-1. Se l’eluente è acqua, 0.1 cm3 min-1 = 5.6 mmol min-1 = 135 cm3 min-1 (c.n.) Nano-HPLC: flussi ~ 10-4 cm3 min-1 0.1 cm3 min (c.n.)
Meccanismo fisico dell’elettrospray L’applicazione di un potenziale elettrico ad alto voltaggio ad una goccia di liquido ne provoca la frammentazione in goccioline fini. La causa e’ la repulsione elettrostatica tra le cariche che si generano nella goccia.
Sistema di nebulizzazione elettrospray Cono di Taylor Punta dell’ago
Sistema di desolvatazione per elettrospray Gocce di circa 1 µm Gas didesolvatazione Gas ditrasporto Liquido ditrasporto Campione Cono di Taylor Capillare riscaldato - + 2-6 kV
Meccanismo di deplezione degli ioni Calore o gas secco Soluzione del campione Spettrometro di massa 4000 V Pressione atmosferica Il calore e/o il gas secco causano la riduzione delle dimensioni delle gocce Vapori del solvente Livelli successivi di vuoto(2 torr – 0.1 torr – 0.01 torr) Esplosione Coulombiana Limite di Rayleigh: Limite di dimensioni della al quale si prevede la frammentazione della goccia. q2 = 8π2ε0γd3 q = carica totale della goccia ε0 = permittività elettrica del mezzo γ = tensione superficiale della goccia d = diametro della goccia
L’elettrospray genera spettri multicarica m/z La carica degli ioni è dovuta agli ioni metallici (es Na+) o ai protoni associati alla molecola. Gli addotti carichi sono quindi della forma: (M + H)+, (M + 2H)2+, …, (M + nH)n+
Sorgenti ioniche a desorbimento Desorbimento/ionizzazione laser assistiti da matrice (Matrix-Assisted Laser Desorption/Ionization, MALDI) • L’analita viene codepositato su un bersaglio con una matrice, scelta per favorire la vaporizzazione dell’analita. • Caratteristiche della matrice: • Elevata assorbività alla lunghezza d’onda del laser. • Massa molare abbastanza bassa da sublimare facilmente. • Bassa reattività chimica. • Stabilità al vuoto. • Le matrici più utilizzate sono acidi aromatici
Il processo MALDI Il bersaglio viene colpito da un impulso laser che viene assorbito dalla matrice e provoca la formazione di un plasma contenente gli atomi dell’analita ed atomi e ioni della matrice.
MALDI-TOF MS Range di massa fino a 106 Analizzatore a tempo di volo.
Lo spettro di massa Intensità relativa m/z Spettro di massa di ioni monocarica (MALDI/TOFMS) Spettro di massa del batterio intero Escherichia coli
MALDI: vantaggi e limitazioni • Poco sensibile alle contaminazioni (sali, tamponi, detergenti ecc.) Solitamente non richiede complessi passaggi di purificazione del campione. • Può analizzare velocemente miscele complesse di analiti (lo spettro è facile da interpretare) • Non è una sorgente a flusso • La ionizzazione è un processo competitivo, per cui la simultanea presenza di più analiti può dare luogo ad interferenze. • L’analisi quantitativa è limitata dalla ridotta omogeneità del campione.
Schema di uno spettrometro di massa Computer Segnale Sistema di introduzione Sorgente di ioni Analizzatore di massa Rivelatore Campione (gassoso) Ioni Ioni 10-5-10-8 torr Campione Sistema di vuoto
Risoluzione La capacità di uno spettrometro di massa di differenziare le masse è generalmente espressa dalla risoluzione R definita come: R = m/Δm dove Δm è la differenza di massa tra due picchi adiacenti risolti e m è la massa nominale del primo picco (o la media delle masse dei due picchi). Due picchi sono considerati separati se l’altezza della valle tra di essi è inferiore ad una certa percentuale della loro altezza (di solito il 10%). Uno spettrometro con una risoluzione di 4 000 risolverà due picchi con valori di m/z 400,0 e 400,1 (o di 40,00 e 40,01). Gli spettrometri commerciali hanno R che variano circa tra 500 e 500 000.
Risoluzione Picchi risolti al 10% della valle Intensità Picchi risolti al 80% della valle Definizione alternativa La risoluzione di un picco isolato si può anche definire come larghezza δm del picco al x% dell’altezza. Spesso si prende x = 50%, e δm è la larghezza a metà altezza.
Classi di analizzatori di massa • A settore magnetico • A doppia focalizzazione • A quadrupolo lineare (Q) • Trappola ionica a quadrupolo (QIT) • A tempo di volo (TOF) • A risonanza ciclotronica e trasformata di Fourier (FT-ICR)
Analizzatore di massa a quadrupolo Potenziale nel quadrupolo
Traiettorie dello ione piano xz piano yz piano xz Traiettoria instabile lungo x, stabile lungo y piano yz Traiettoria stabile sia lungo x che lungo y
Traiettorie ioniche nella trappola a quadrupolo Moto degli ioni in direzione z Moto degli ioni in direzione r tempo Gli ioni vengono intrappolati forzandoli su traiettorie chiuse stabili all’interno della trappola. Quando la traiettoria di uno ione diventa instabile, questo esce dalla trappola e viene rivelato.
Analizzatore di massa a quadrupolo • Massimo valore m/z ~ 4 000 • Risoluzione ~ 3 000 • I quadrupoli sono strumenti a bassa risoluzione • Si lavora normalmente alla risoluzione di una unità di massa. • Leggero, di dimensioni contenute • Facile da accoppiare alla cromatografia • Efficiente trasmissione degli ioni • Necessaria una elevata precisione nell’allineamento delle barre degli elettrodi
Analizzatore a tempo di volo (TOF) Zona di accelerazione con campo elettrostatico E Ioni positivi Sorgente d V = 0 dE = V0 = 20 000 V V = V0 L
Analizzatore di massa a tempo di volo • Massimo valore m/z > 200 000 • Risoluzione fino a 105. • Il reflectron consente di: • Ridurre le dimensioni • Aumentare la risoluzione • Ideale accoppiamento con sorgenti pulsate • Efficiente trasmissione degli ioni • Per la doppia spettrometria di massa può essere accoppiato con altri analizzatori (qTOF)
Principi della ICRFT MS Gli ioni in un campo elettromagnetico si muovono su traiettorie circolari (o a spirale), generando esse stesse una radiazione elettromagnetica. Tale r.e.m. può essere rivelata da un’antenna, generando un segnale che contiene le radiofrequenze di tutti gli ioni in funzione del tempo. Mediante trasformata di Fourier il segnale viene passato dal dominio del tempo al dominio delle frequenze, e di conseguenza al dominio delle masse.
Analizzatore a risonanza ciclotronica Piano di ricezione Piano di emissione
Analisi di massa a trasformata di Fourier Segnale nel dominio del tempo Segnale trasformato nel dominiodelle frequenze m/z
Analizzatore di massa a ICR FT • Massimo valore m/z > 50 000 • Risoluzione fino a 106. • Possibilità di confinare gli ioni per la MS/MS. • Massima accuratezza nella determinazione delle masse. • Semplicità nel passaggio da ioni positivi a ioni negativi. • Prezzi ancora proibitivi per molte applicazioni (500 –1 400 k€)
Doppia spettrometria di massa (MS/MS) Ionizzazione eframmentazione analisidi massa m1 analisidi massa m1 decomposizione m/z m/z
Applicazioni della MS/MS • Maggior contenuto di informazioni • Studio della struttura • Informazioni addizionali per determinare la struttura di uno ione • Rivelazione selettiva di uno ione • Drastica riduzione delle interferenze • Studio delle reazioni ione-molecola
MS/MS nello spazio e nel tempo Cella dicollisione MS/MS nello spazio Tempo 1 Tempo 2 Tempo 3 MS/MS nel tempo Scansione delloione prodotto Selezione m/z Scansione
Combinazioni strumentali per MS/MS • MS/MS nello spazio • Settori elettrico-magnetico: EB/EB o BE/BE (max MS4). • Triplo quadrupolo: QqQ • Tempo di volo lineare/reflectron: TOF/TOF. • Analizzatori ibridi: EBqQ • QqTOF • MS/MS nel tempo (MSn) • Trappola ionica a quadrupolo (max MS8) • Risonanza ciclotronica (ICR FTMS) • Combinati: Qq(trap) MSn nello spazio e/o nel tempo
Modalità di scansione MS/MS Analizzatore di massa fisso Spettrometro di massa a scansione Scansione dello ione prodotto Scansione dello ione precursore Scansione di perdita neutra Monitoraggio di reazione selezionata Scansione dello ione prodotto Selezione m/z Scansione Simbolismo alternativo Scansione dello ione precursore Selezione m/z Scansione Scansione di perdita neutra Scansionem/z = x Scansionem/z = x-a Monitoraggio di reazione selezionata Selezione delframmentom/z = b Selezione delprecursorem/z = a
Modalità di scansione MS/MS • Scansione dello ione prodotto: consiste nel selezionare uno ione precursore, di m/z fissato, e nella determinazione di tutti gli ioni prodotti dalla frammentazione attivata per collisione (CID).Se nella cella di collisione si usa un gas reagente, si osservano i prodotti di reazione attivate per collisione (CAR). Quando vengono prodotti solo ioni frammento, questa modalità di scansione è detta anche scansione di ioni frammento. • Scansione dello ione precursore: consiste nello scegliere uno ione prodotto e nel determinare gli ioni precursori. Permette di identificare gli ioni precursori di un certo frammento. Questa modalità di scansione, che non si può realizzare con la in time MS/MS, richiede la focalizzazione del secondo analizzatore su di uno ione selezionato, e la scansione di massa sul primo analizzatore. Vengono rivelati gli ioni precursori che per reazione o frammentazione producono ioni figlio della massa selezionata.
Modalità di scansione MS/MS • Scansione di perdita neutra (NLS): consiste nello scegliere un frammento neutro e nel rivelare tutte le frammentazioni che portano alla perdita di tale frammento. Come nel caso della scansione dello ione precursore, questa modalità non è realizzabile in MS/MS nel tempo. La scansione richiede che entrambi gli analizzatori scandiscano contemporaneamente, ma con una differenza di massa fissa a fra di loro. Quando uno ione di massa m attraversa il primo analizzatore, viene rivelato solo se produce un frammento di massa m – a. • Monitoraggio di reazioni selezionate (SRM): consiste nel selezionare una reazione di frammentazione. In questa modalità, entrambi gli analizzatori sono focalizzati su masse selezionate. Non si ha quindi scansione. È analogo al “monitoraggio di ioni selezionati” in spettrometria di massa singola, ma in questo caso gli ioni selezionati dal primo analizzatore danno un segnale solo se producono un certo frammento mediante una certa reazione. L’assenza di scansione permette di aumentare la sensibilità, oltre che la selettività.
Combinazioni strumentali per MS/MS • MS/MS con risoluzione nello spazio • Settori elettrico-magnetico: EB/EB o BE/BE (max MS4). • Triplo quadrupolo: QqQ • Tempo di volo lineare/reflectron: TOF/TOF. • Combinati: EBqQ • QqTOF • MS/MS con risoluzione nel tempo (MSn) • Trappola ionica a quadrupolo (max MS8) • Risonanza ciclotronica (ICR FTMS) • Combinati: Qq(trap) MSn nello spazio e/o nel tempo
MS di molecole biologiche • Classi di biomolecole • Proteine - Peptidi • Acidi nucleici • Polisaccaridi • Lipidi • Tecniche MS • Sorgenti: (FAB), MALDI, ESI • Analizzatori: TOF, quadrupolo (con ESI), FT ICR, ibridi • Tecniche separative • HPLC, SEC, CZE, elettroforesi su gel.
I progetti -OMICI • Genoma • Identificare tutta la serie di ordini che il codice genetico può impartire ad una cellula, per comprendere e prevenire situazioni patologiche • Trascrittoma • Identificare e quantificare tutta la serie di ordini che il genoma impartisce effettivamente alla cellula, per comprendere ed eventualmente correggere le modalità di regolazione genica • Proteoma • Caratterizzare e quantificare il contenuto proteico di un sistema cellulare, mediante la determinazione di profili di espressione differenziati nel tempo e nello spazio, comprensivi delle informazioni sulle interazioni molecolari
Massa monoisotopica e massa chimica Massa monoisotopica (picco 12C): 1085.55 Massa media (picco 12C): 1086.28 Per peptidi e proteine la differenza fra massa media (chimica) e massa mono-isotopica è circa 0.06%, distinguibile se lo spettrometro ha risoluzione pari a: R = 100/0.06 = 1667 (nell’esempio sopra, R = 5000)
Identificazione del picco 12C Insulina Albumina bovina Per piccoli peptidi il primo picco, relativo alla massa monoisotopica, è anche quello più intenso. Questo non è vero per proteine di massa maggiore, in cui il picco 12C può essere poco intenso o addirittura non rivelabile. Oltre un certo valore di massa, il dato analitico utile è la massa media, che si ricava dal centroide del picco.
Risoluzione e sensibilità Quando non è possibile raggiungere risoluzione unitaria, ed identificare il picco di massa monoisotopica, è poco conveniente superare la risoluzione alla quale si può determinare la massa media con accuratezza. Questo è vero in particolare quando si deve raggiungere un compromesso fra risoluzione e sensibilità
Ruolo della MS in proteomica • Proteomica sistematica • Identificazione e sequenziamento di tutte le proteine contenute in una cellula. (MS o MS/MS accoppiata a 2D PAGE o 2D LC per l’analisi strutturale di miscele di peptidi e proteine intere) • Proteomica differenziale • Studio delle differenze rilevabili fra il proteoma di una cellula sana e quello di una cellula malata (2D PAGE + MS o MS/MS per la determinazione di mutazioni nella sequenza, modifiche post-traduzionali, modifiche conformazionali, sovra o sottoespressione) • Proteomica funzionale • Studio della funzione biologica delle proteine e delle loro interazioni con proteine e peptidi (Separazione in condizioni non denaturanti + MS di proteine e complessi proteici allo stato nativo)
Strategie proteomiche basate sulla MS Proteina (sconosciuta) bottom-up 2D SDS PAGE Top-down digestione Miscela di peptidi Massa dei peptidi mediante LC/LC/ESIMS shotgun Ricerca dei pesi molecolari in banca dati Massa di proteine intere mediante (LC)ESI/MS sconosciuta nota Sequenziamento in MSn sconosciuta MSn per ulteriori Informazioni sulla sequenza nota Identificazione e caratterizzazione di proteine Ricerca dei pesi molecolari in banca dati
Approccio proteomico top-down • Si basa sulla determinazione della massa accurata (media) delle proteine in una matrice di complessità ridotta (ottenuta per purificazione con metodi biochimici o per separazione con tecniche non denaturanti) • Permette di determinare la conformazione delle proteine nel loro stato nativo, e le interazioni proteina-proteina (complessi proteici fra biomarker e proteine di trasporto, oligomeri funzionali) • Permette di determinare la composizione amminoacidica della proteina, ma non la loro sequenza • Non identifica la proteina in modo univoco in un proteoma complesso • Permette di studiare isoforme (mutazioni di singoli amminoacidi) e modificazioni post-traduzionali, ma non di localizzarle.
Approccio proteomico top-down Spettro m/z in ESI/qTOF di perossidasidi rafano (HRP) allo stato nativo Il software dello spettrometro di massa identifica le famiglie di picchi negli spettri multicarica sovrapposti, ne determina il valore di m/z medio (il centroide del picco) e calcola lo spettro di massa ricostruito
Approccio proteomico top-down F 43243.0 2Ca2+ 2Ca2+ D 42422.0 E 43165.0 C 42343.1 2Ca2+ A 41521.9 B 41600.4 Glicosilazione Glicosilazione Sullo spettro di massa ricostruito si possono identificare isoforme, complessi metallo-proteina e modificazioni post-traduzionali
LC-MS/MS per l’identificazione “de novo” della sequenza peptidica MS/MS MS y5 y9 y6 b8 b9 b11 y3 y7 y4 y10 b7 b3 b12 y2 m/z 200 600 1000 MS trace MS/MS trace 30 40 50 Time [min]
Frammentazione dei peptidi x1 y1 z1 x2 y2 z2 x3 y3 z3 R4 R3 R2 R1 O O O O NH CH-COH C NH CH C NH CH C H2N-CH a1 b1 c1 a2 b2 c2 a3 b3 c3 R4 R3 + R2 + R1 O O O O NH CH-COH C NH3 CH C NH CH C H2N-CH b2 y”2