Download

1 / 27
280 likes | 498 Views
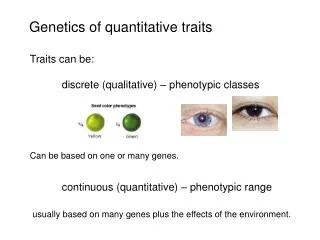
Mapping of complex traits Andy Willaert Center for Medical Genetics Ghent. Complex traits. Complex traits: Diabetes, Crohn, Hypertension, Osteoporosis,... Complex inheritance patterns Many different gene-variants involved each having a small effect! Importance of environmental factors!

E N D
Mapping of complex traits Andy Willaert Center for Medical Genetics Ghent
Complex traits Complex traits: Diabetes, Crohn, Hypertension, Osteoporosis,... Complex inheritance patterns • Many different gene-variants involved each having a small effect! • Importance of environmental factors! • Importance of gene-gene interactions and gene-environment interactions! Traditional linkage analyses difficult for complex traits Gausse curve
Mapping of complex traits • Model-based linkage analysis (parametric): • Depends on knowing that a mutation in a single gene is inherited in a specific mendelian inheritance pattern • Powerful method for mapping single-gene disorders • Not very useful for complex traits • Model-free linkage analysis (non-parametric): • Does not assume any particular mode of inheritance to explain the inheritance pattern • Depends solely on the assumption that affected relatives will be more likely to have disease-predisposing alleles in common than is expected by chance.
Affected sibpair method • Affected sibpair method In general: • Relies on pairs of family members such as siblings, concordant for the phenotype • Siblings have on average one allele of two in common at any locus (Full siblings share on average 50% of their DNA) • If an allele is shared more frequently than expected (more than 50%) by sibs concordant for a particular phenotype, than the allele predisposes to that phenotype
Affected sibpair method • Affected sibpair method In practice: • DNA of a set of affected sibs or affected individuals in families is analysed by use of hundreds of polymorphic markers throughout the entire genome (genome scan) • Elevated degrees of allele sharing (significantly more than 50 %) between affected pairs at a polymorphic marker suggests that a locus involved in the disease is located close to the marker • Degree of allele-sharing can be assessed by use of a non-parametric LOD-score (NPL-score) which is comparable to parametric LOD-score • NPL-score >3.6 = evidence for increased allele-sharing NPL-score >5.4 = highly significant increased allele-sharing
Affected sibpair method Affected sibpair method does not require to make assumptions about the inheritance patterns, but method is rather insensitive and imprecise • Insensitivity is reflected in the fact that large numbers of sibpairs or relatives are required to detect a significant deviation from the expected 50% allele-sharing – many hundreds/thousands of sibpairs or families needed • Imprecise: Only broad regions of increased allele-sharing can be identified and not a narrow, critical interval as in model-based linkage analysis
Association analysis Association analysis • Analysis of the DNA of two groups of participants: people with the disease being studied and similar people without the disease. • If certain genetic variations are found more frequently in people with the disease compared to people without disease, the variations are said to be "associated" with the disease.
Association analysis • The strength of an association between disease and genotype is calculated by an odds ratio Disease Odds Ratio for allele A = the chance that an allele A carrier develops the disease divided by the chance that an allele A non- carrier develops the disease a Disease Odds Ratio for allele A = b = ad = 23X116 = 6.9 c bc 4X97 d !! Seven times higher chance of getting the disease if a person carries the allele A than if the person carries the B allele
Association analysis • The significance of an association can be assessed by performing χ2 test: Test if values of a, b, c and d differ from what would be expected if there was no association Χ2 = 15 with 1 df; P < 10-10 Highly significant association between allele A and the disease!
Association analysis • Strengths association studies: • Powerful tool for pinpointing precisely the genes and the alleles that contribute to genetic disease • No need to carry out laborious family studies and collection of samples from many members of a pedigree • Weaknesses association studies: • Population stratification: - A disease that happens to be more common in a certain subpopulation and any allele that also happens to be more common in that certain subpopulation can be falsely associated. - Can be avoided by careful selection of cases and controls (not sampled from different subpopulations) or by using family-based association study designs
Association analysis • Weaknesses association studies: • Linkage disequilibrium (LD): - All alleles in LD with an allele involved in the disease will show an apparently positive association whether they have any functional relevance in disease predispotion or not - Still useful, since the associated alleles must at least be in loci that are close enough to the real disease locus to appear associated LD1 LD2 AT A G C
Genome-wide Association analysis • Genome-wide association (GWA)studies • Until recently, association studies have been limited to particular sets of variants in restricted sets of genes • Recently more powerful genome-wide association studies are being performed, without any preconception of what genes and genetic variants migth be contributing to the disease
Genome-wide Association analysis • What has made genome-wide association possible? • Publication of the sequence of the human genome in 2001. This sequence has been very informative about the vast majority of bases that are invariant across individuals. • HAPMAP project focuses on DNA sequence differences among individuals → SNPs were characterised in 270 individuals in four different populations: European, African, Chinese and Japanese populations and a first map of 1.3 million common SNPs was published in 2005, extended to 3.1 million SNPs in 2007. LD-patterns between SNPs revealed. • Genome-wide association studies require the ability to genotype a sufficiently set of variants in a large patient sample for a low cost: High throughput genotyping platforms available: Affymetrix/Illumina chips
Genome-wide Association analysis • Tagging SNPs for genome-wide association • Hapmap provides information about LD between SNPs on the genome and divides the genome into LD-blocks of about 10 kb in European population Restricted number of haplotypes within LD block Tagging SNPs capture most frequent haplotypes • Genotyping a few hundred thousand tag SNPs in a GWA-study only a bit less useful than genotyping all 10 million common SNPs
Genome-wide Association analysis • A Catalog of Published Genome-Wide Association Studieshttp://www.genome.gov/gwastudies/
Nature of genetic component contributing to complex traits? • ‘Common Disease, Common Variant (CDCV)’ hypothesis: genetic variations with relatively high frequency in the population, but relatively low penetrance, are the major contributors tot genetic susceptibility to common diseases. • But: Genetic variants from GWA: explain only small fraction (5%) of heritable risk for common diseases • ’Common Disease, Rare Variant (CDRV)’ hypothesis: multiple rare DNA sequence variations, each with relatively high penetrance, are the major contributors to genetic susceptibility to common diseases CDCV versus CDRV
Case studies Case studies Positional cloning: the overall strategy of mapping the location of a disease gene by linkage/associaton, followed by attempts to identify the gene on the basis of its map position.
Case studies • Positional cloning of a complex disease by genome-wide association: Age-related Macular Degeneration (AMD) • Progressive degenerative disease of the portion of the retina, responsible for central vision causing blindness in 1.75 million Americans older than 50y • Characterised by the accumulation of extracellular protein behind the retina in the region of the macula • Ample evidence for a genetic contribution, although most AMD patients are not in families with a clear mendelian pattern • Environmental contributions important (increased risk of AMD in cigarette smokers)
Case studies • Positional cloning of a complex disease by genome-wide association: Age-related Macular Degeneration (AMD) • Case (96) –control (50) genome-wide association study using 116.000 SNPs revealed association of alleles at two common SNPs with AMD. • Both alleles showed an odds ratio of 4 and 7 in affected individuals who were respectively heterozygous and homozygous for either of these alleles. • Both SNPs were located within an intron of the gene encoding complement factor H (CFH), important in inflammation • Examination of the HAPMAP revealed that these two SNPs were in LD with SNPs across a 41 kB LD-block on chromosome 1 • Search through the SNPs in the 41 kb LD-block revealed a nonsynonymous SNP (Tyr402His) in the CFH gene, with even stronger association with AMD
Case studies • Positional cloning of a complex disease by genome-wide association: Age-related Macular Degeneration (AMD) • Replication in other case-control samples with AMD and estimated to be responsible for 43% of all the genetic contribution to the disease • CFH protein is found in retinal tissue, protecting against inflammation and the resulting accumulation of extracellular protein. The Tyr402His variant of the CFH gene is less protective! • Consequently, variants in other components of the complement system have been investigated as candidate loci for AMD: SNPs in factor B and complement factor 2, altering amino acids, are associated with AMD. • Conclusion: For the complex disorder AMD, a genome-wide association study finally led to the identification of SNPs at CFH, complement factor 2 and factor B, estimated to account for most of the genetic contribution to AMD.
Case studies • Chronic inflamatory disease of the gastrointestinal tract that primarily affects adolescents and young adults • Divided into two major categories: Crohn disease and ulcerative colitis (UC) • Family and Twin studies provided ample evidence for a genetic contribution to Crohn, although most patients are not in families with a clear mendelian pattern • Positional cloning of a complex disease by model-free Linkage mapping: Inflammatory Bowel Disease (Crohn)
Case studies • Positional cloning of a complex disease by model-free Linkage mapping: Inflammatory Bowel Disease (Crohn) • Many genome scans using model-free linkage analysis carried out in families with two or more IBD affected individuals • 11 genomic regions with positive NPL scores, the one with the highest score (>5,4) showing linkage to Crohn only and not to UC (most of the other regions showed linkage to both forms of IBD) • A locus, termed IBD1, was proposed to reside in this region (16q12) of the highest LOD-score • Association study using SNPs in the region of 160 kb around the marker with the highest NPL score revealed three SNPs with strong evidence for LD with the disease. • Three SNPs located in the coding exons for the gene NOD2 or CARD15, causing either amino acid substitutions (Arg702Trp, Gly908Arg) or premature protein termination(Leu1007fsinsC)
Case studies • Positional cloning of a complex disease by model-free Linkage mapping: Inflammatory Bowel Disease (Crohn) • NOD2 protein binds to gram-negative bacterial cell walls and participates in the inflammatory response to bacteria by activating NF-kB transcription factor in mononuclear leukocytes • The three variants reduce the ability of NOD2 to activate NF-kB, altering the ability of monocytes in intestinal wall to respond to resident bacteria, predisposing to an abnormal inflammatory response • Additional association studies in several independent cohorts of Crohn patients confirmed strong association of the three variants with Crohn • Genetic contribution of NOD2 variants is supported by a dosage effect: -Heterozygotes for NOD2 variants have odds ratio of 1.5 to 4 -Homozygotes for NOD2 variants have odds ratio of 15 to 40
Case studies • Positional cloning of a complex disease by model-free Linkage mapping: Inflammatory Bowel Disease (Crohn) • Discovery of NOD2 variants helps explain complex inheritance pattern in Crohn: 1) Three NOD2 variants not necessary to cause Crohn • Half of all white patients with Crohn disease have one or two copies of a NOD2 variant, half do not. • Three NOD2 variants are associated with Crohn in Europe, but are not found in Asian or African populations (NOD2 is not associated with Crohn in these populations) 2) Three NOD2 variants not sufficient to cause Crohn • 20 % of the European population is heterozygous for the three variants and show no signs of Crohn • Homozygotes and compound heterozygotes for the NOD2 variants show penetrance less than 10%
Case studies • Positional cloning of a complex disease by model-free Linkage mapping: Inflammatory Bowel Disease (Crohn) • Conclusions: 1) Other genetic or environmental factors acting on the genotypic susceptibility at the NOD2 locus 2) The obvious connection between Crohn disease (inflammatory bowel disease) and structural variants in NOD2 (modulator of antibacterial inflammatory response) is a strong clue to what some of these other genetic /environmental factors might be 3) Genetic analysis of Crohn disease exemplifies how to think about complex traits and how to identify genetic contributions