Download
1 / 94
940 likes | 1.03k Views
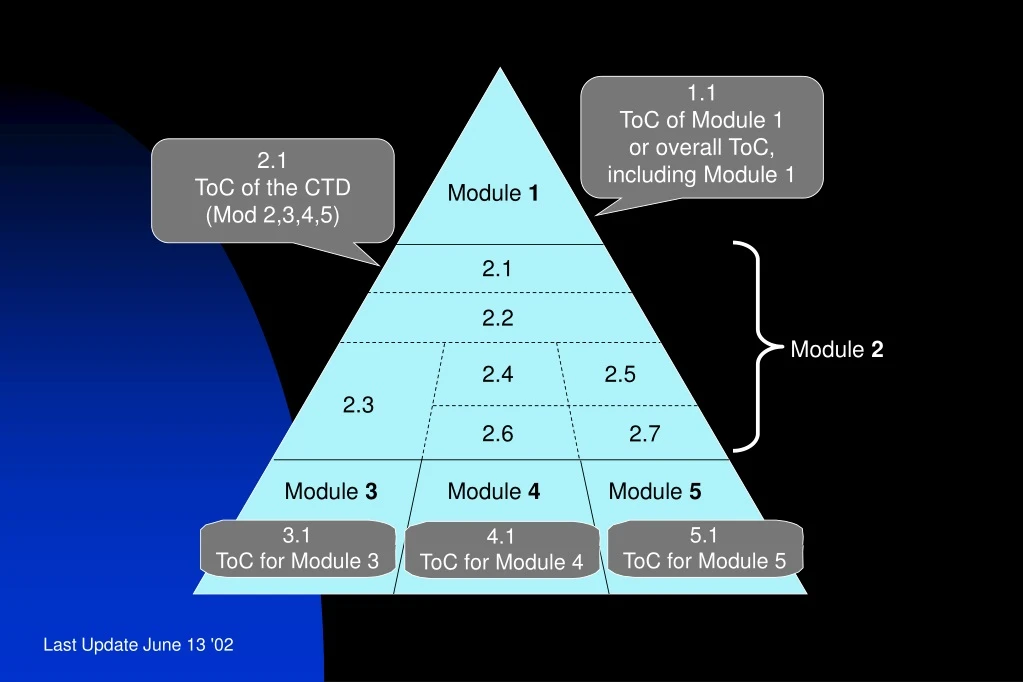
1.1 ToC of Module 1 or overall ToC, including Module 1. 2.1 ToC of the CTD (Mod 2,3,4,5). Module 1. 2.1. 2.2. Module 2. 2.4. 2.5. 2.3. 2.6. 2.7. Module 3. Module 4. Module 5. 3.1 ToC for Module 3. 5.1 ToC for Module 5. 4.1 ToC for Module 4.

E N D
1.1 ToC of Module 1 or overall ToC, including Module 1 2.1 ToC of the CTD (Mod 2,3,4,5) Module 1 2.1 2.2 Module 2 2.4 2.5 2.3 2.6 2.7 Module 3 Module 4 Module 5 3.1 ToC for Module 3 5.1 ToC for Module 5 4.1 ToC for Module 4
1.0 Regional Administrative Information 1.1 ToC of Module 1 or overall ToC, including Module 1 Module 1 1.0 2.1 ToC of the CTD (Mod 2,3,4,5) 2.2 Introduction 2.1 Module 2 2.3 Quality Overall Summary 2.2 2.4 Nonclinical Overview 2.4 2.5 2.5 Clinical Overview 2.3 2.6 Nonclinical Written and Tabulated Summaries 2.6 2.7 Module 3 Module 4 Module 5 2.7 Clinical Summary Clinical Study Reports Nonclinical Study Reports Quality 4) Numbering System
4.1 Table of Contents of Module 4 • A Table of Contents should be provided that lists all of the nonclinical study reports and gives the location of each study report in the Common Technical Document.
4.2 Study Reports • The study reports should be presented in the following order: • 4.2.1 Pharmacology • 4.2.1.1 Primary Pharmacodynamics • 4.2.1.2 Secondary Pharmacodynamics • 4.2.1.3 Safety Pharmacology • 4.2.1.4 Pharmacodynamic Drug Interactions
Species should be ordered as follows: • • Mouse • • Rat • • Hamster • • Other rodent • • Rabbit • • Dog • • Non-human primate • • Other non-rodent mammal • • Non-mammals
Routes of administration should be ordered as follows : • • The intended route for human use • • Oral • • Intravenous • • Intramuscular • • Intraperitoneal • • Subcutaneous • • Inhalation • • Topical • • Other
4.2.2 Pharmacokinetics • 4.2.2.1 Analytical Methods and Validation Reports (if separate reports are available) • 4.2.2.2 Absorption • 4.2.2.3 Distribution • 4.2.2.4 Metabolism • 4 2.2.5 Excretion • 4.2.2.6 Pharmacokinetic Drug Interactions (nonclinical) • 4.2.2.7 Other Pharmacokinetic Studies
4.2.3 Toxicology • 4.2.3.1 Single-Dose Toxicity (in order by species, by route) • 4.2.3.2 Repeat-Dose Toxicity (in order by species, by route, by duration; including supportive toxicokinetics evaluations) • 4.2.3.3 Genotoxicity • 4.2.3.3.1 In vitro • 4.2.3.3.2 In vivo (including supportive toxicokinetics evaluations)
4.2.3 Toxicology • 4.2.3.4 Carcinogenicity (including supportive toxicokinetics evaluations) • 4.2.3.4.1 Long-term studies (in order by species; including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) • 4.2.3.4.2 Short- or medium-term studies (including range-finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics) • 4.2.3.4.3 Other studies
4.2.3 Toxicology • 4.2.3.5 Reproductive and Developmental Toxicity (including range-finding studies and supportive toxicokinetics evaluations) (If modified study designs are used, the following sub-headings should be modified accordingly.) • 4.2.3.5.1 Fertility and early embryonic development • 4.2.3.5.2 Embryo-fetal development • 4.2.3.5.3 Prenatal and postnatal development, including maternal function • 4.2.3.5.4 Studies in which the offspring (juvenile animals) are dosed and/or further evaluated.
4.2.3 Toxicology • 4.2.3.6 Local Tolerance • 4.2.3.7 Other Toxicity Studies (if available) • 4.2.3.7.1 Antigenicity • 4.2.3.7.2 Immunotoxicity • 4.2.3.7.3 Mechanistic studies (if not included elsewhere) • 4.2.3.7.4 Dependence • 4.2.3.7.5 Metabolites • 4.2.3.7.6 Impurities • 4.2.3.7.7 Other
5.1 Table of Contents of Module 5 • A Table of Contents for study reports should be provided. 5.2 Tabular Listing of All Clinical Studies
5.3 Clinical Study Reports • 5.3.1 Reports of Biopharmaceutic Studies • 5.3.1.1 Bioavailability (BA) Study Reports • BA studies in this section should include • studies comparing the release and systemic availability of a drug substance from a solid oral dosage form to the systemic availability of the drug substance given intravenously or as an oral liquid dosage form • • dosage form proportionality studies, and • • food-effect studies.
5.3.1.2 Comparative BA and Bioequivalence (BE) Study Reports • Comparative BA or BE studies may include comparisons between : • the drug product used in clinical studies supporting effectiveness and the to-be-marketed drug product, • • the drug product used in clinical studies supporting effectiveness and the drug product used in stability batches, and • • similar drug products from different manufacturers.
5.3.1.3 In vitro-In vivo Correlation Study Reports • 5.3.1.4 Reports of Bioanalytical and Analytical Methods for Human Studies
5.3 Clinical Study Reports • 5.3.2 Reports of Studies Pertinent to Pharmacokinetics using Human Biomaterials • 5.3.2.1 Plasma Protein Binding Study Reports • 5.3.2.2 Reports of Hepatic Metabolism and Drug Interaction Studies • 5.3.2.3 Reports of Studies Using Other Human Biomaterials • 5.3.3 Reports of Human Pharmacokinetic (PK) Studies • 5.3.3.1 Healthy Subject PK and Initial Tolerability Study Reports • 5.3.3.2 Patient PK and Initial Tolerability Study Reports • 5.3.3.3 Intrinsic Factor PK Study Reports • 5.3.3.4 Extrinsic Factor PK Study Reports • 5.3.3.5 Population PK Study Reports
5.3 Clinical Study Reports • 5.3.4 Reports of Human Pharmacodynamic (PD) Studies • 5.3.4.1 Healthy Subject PD and PK/PD Study Reports • 5.3.4.2 Patient PD and PK/PD Study Reports • 5.3.5 Reports of Efficacy and Safety Studies • 5.3.5.1 Study Reports of Controlled Clinical Studies Pertinent to the Claimed Indication • 5.3.5.2 Study Reports of Uncontrolled Clinical Studies • 5.3.5.3 Reports of Analyses of Data from More Than One Study • 5.3.5.4 Other Clinical Study Reports
5.3.5.1 Study Reports of Controlled Clinical Studies Pertinent to the Claimed Indication • The controlled clinical study reports should be sequenced by type of control: • Placebo control (could include other control groups, such as an active comparator or other doses) • No-treatment control • Dose-response (without placebo) • Active control (without placebo) • External (Historical) control, regardless of the control treatment
5.3.5.4 Other Clinical Study Reports • Reports of interim analyses of studies pertinent to the claimed indications • − Reports of controlled safety studies not reported elsewhere • − Reports of controlled or uncontrolled studies not related to the claimed indication • Published reports of clinical experiences with the medicinal product that are not included in Section 5.3.5.1. However, when literature is important to the demonstration or substantiation of efficacy, it should be included in Section 5.3.5.1 • − Reports of ongoing studies
5.3 Clinical Study Reports • 5.3.6 Reports of Post-Marketing Experience • 5.3.7 Case Report Forms and Individual Patient Listings
THE EXTENT OF POPULATION EXPOSURE • TO ASSESS CLINICAL SAFETY • FOR DRUGS INTENDED FOR LONG-TERM TREATMENT OF • NON-LIFE-THREATENING CONDITIONS • E1
The objective of this guideline is to present an accepted set of principles for the safety evaluation of drugs intended for the long-term treatment (chronic or repeated intermittent use for longer than 6 months) of non-life-threatening diseases. The safety evaluation during clinical drug development is expected to characterise and quantify the safety profile of a drug over a reasonable duration of time consistent with the intended long-term use of the drug. Thus, duration of drug exposure and its relationship to both time and magnitude of occurrence of adverse events are important considerations in determining the size of the data base necessary to achieve such goals.
CLINICAL SAFETY DATA MANAGEMENT: • DEFINITIONS AND STANDARDS FOR • EXPEDITED REPORTING • E2A
There are two issues within the broad subject of clinical safety data management that are appropriate for harmonisation at this time: • (1) the development of standard definitions and terminology for key aspects of clinical safety reporting, and • (2) the appropriate mechanism for handling expedited (rapid) reporting, in the investigational (i.e., pre-approval) phase.
KEY DATA ELEMENTS FOR INCLUSION IN EXPEDITED • REPORTS OF SERIOUS ADVERSE DRUG REACTIONS
1. Patient Details • Initials • Other relevant identifier (clinical investigation number, for example) • Gender • Age and/or date of birth • Weight • Height
2. Suspected Medicinal Product(s) • Brand name as reported • International Non-Proprietary Name (INN) • Batch number • Indication(s) for which suspect medicinal product was prescribed or tested • Dosage form and strength • Daily dose and regimen (specify units - e.g., mg, ml, mg/kg) • Route of administration • Starting date and time of day • Stopping date and time, or duration of treatment
Details of Suspected Adverse Drug Reaction(s) • Full description of reaction(s) including body site and severity, as well as the criterion (or criteria) for regarding the report as serious should be given. In addition to a description of the reported signs and symptoms, whenever possible, attempts should be made to establish a specific diagnosis for the reaction. • Start date (and time) of onset of reaction • Stop date (and time) or duration of reaction • Dechallenge and rechallenge information • Setting (e.g., hospital, out-patient clinic, home, nursing home) • Outcome:
Details on Reporter of Event (Suspected ADR) • Name • Address • Telephone number • Profession (speciality) • Administrative and Sponsor/Company Details
CLINICAL SAFETY DATA MANAGEMENT: • PERIODIC SAFETY UPDATE REPORTS FOR MARKETED DRUGS • E2C(R1)
The current situation for periodic safety reports on marketed drugs is different among the three ICH regions. For example: • − The US regulations require quarterly reports during the first 3 years, then annual reports. The FDA has recently published proposed rules1 which take into account the CIOMS Working Group II proposals2. • − In the EU, Council Directive 93/39/EEC and Council Regulation 2309/93 require reports with a periodicity of 6 months for two years, annually for the three following years and then every five years, at time of renewal of registration. • − In Japan, the authorities require a survey on a cohort of a few thousand patients established by a certain number of identified institutions during the 6 years following authorization. Systematic information on this cohort, taking into account a precise denominator, must be reported annually. Regarding other marketing experience, adverse reactions which are non-serious, but both mild in severity and unlabeled must be reported every 6 months for 3 years and annually thereafter.
SAMPLE TITLE PAGE • PERIODIC SAFETY UPDATE REPORT FOR: (PRODUCT) • MAHs NAME AND ADDRESS (Corporate headquarters or other company entity responsible for report preparation) • PERIOD COVERED BY THIS REPORT: (dates) • INTERNATIONAL BIRTH DATE: Date (Country of IBD) • DATE OF REPORT • (Other identifying information at the option of MAH, such as report number)
TABLE OF CONTENTS FOR MODEL PSUR • Introduction ......................................................................................................... • World-wide market authorization status........................................................................... • Update of regulatory authority or MAH actions taken for safety reasons ..................... • Changes to Reference Product Information....................................................................... • Patient exposure.................................................................................................................. • Presentation of individual case histories............................................................................ • Studies ....................................................................................................................... • Other information................................................................................................................ • Overall safety evaluation.................................................................................................... • Conclusion ....................................................................................................................... • APPENDIX: COMPANY CORE DATA SHEET.................................................................
3. GLOSSARY OF SPECIAL TERMS • Company Core Data Sheet (CCDS) • A document prepared by the MAH containing, in addition to safety information, material relating to indications, dosing, pharmacology and other information concerning the product.
Company Core Safety Information (CCSI) • Data Lock-Point (Data Cut-off Date) • International Birth Date IBD • Listed Adverse Drug Reaction • Spontaneous Report or Spontaneous Notification • Unlisted Adverse Drug Reaction
ADDENDUM TO ICH E2C • CLINICAL SAFETY DATA MANAGEMENT PERIODIC SAFETY UPDATE REPORTS FOR MARKETED DRUGS • ICH Harmonised Tripartite Guideline
Summary Bridging Report • Addendum Report • Proprietary information • Executive Summary • Risk management programme • Benefit-risk analysis
POST-APPROVAL SAFETY DATA MANAGEMENT: DEFINITIONS AND STANDARDS FOR EXPEDITED REPORTING • E2D
PHARMACOVIGILANCE PLANNING • E2E • This guideline is intended to aid in planning pharmacovigilance activities, especially in preparation for the early postmarketing period of a new drug • The guideline describes a method for summarising the important identified risks of a drug, important potential risks, and important missing information, including the potentially at-risk populations and situations where the product is likely to be used that have not been studied pre-approval.
1. TITLE PAGE • The title page should contain the following information: • − study title • − name of test drug/ investigational product • − indication studied • − if not apparent from the title, a brief (1 to 2 sentences) description giving design (parallel, cross-over, blinding, randomised) comparison (placebo, active, dose/response), duration, dose, and patient population • − name of the sponsor • − protocol identification (code or number) • − development phase of study • − study initiation date (first patient enrolled, or any other verifiable definition) • − date of early study termination, if any • − study completion date (last patient completed) • − name and affiliation of principal or coordinating investigator(s) or sponsor’s responsible medical officer • − name of company/sponsor signatory (the person responsible for the study report within the company/sponsor. The name, telephone number and fax number of the company/sponsor contact persons for questions arising during review of the study report should be indicated on this page or in the letter of application.) • − statement indicating whether the study was performed in compliance with Good Clinical Practices (GCP), including the archiving of essential documents • − date of the report (identify any earlier reports from the same study by title and date).