Download

1 / 39
390 likes | 511 Views
Framework for Sequence Cluster Merging ( Also showing importance of domain knowledge ). Arvind Gopu Masters student, Computer Science & Bioinformatics Indiana University, Bloomington http://biokdd.informatics.indiana.edu/~agopu Email: agopu@cs.indiana.edu. Introduction.

E N D
Framework for Sequence Cluster Merging (Also showing importance of domain knowledge) Arvind Gopu Masters student, Computer Science & Bioinformatics Indiana University, Bloomington http://biokdd.informatics.indiana.edu/~agopu Email: agopu@cs.indiana.edu
Introduction • Sequence Clustering very important research topic. • Bottom-up approach – basically merge elements recursively upto certain specificity • Top-down approach – split elements until desired specificity is achieved • Two important issues: selectivity and sensitivity • Sequence clustering problem is unique • No “observable” attributes unlike most clustering problems • Example: • Supermarket: Soda, Fruit juice, Frozen foods, Clothing, etc. • Demographic: Height, Race, etc. • Sequence clustering: Just a bunch of amino acid characters! (with accompanying well studied sequence comparison/alignment programs).
Introduction … • Getting back to sequence clustering… • Fragmentation problem – well known in sequence clustering algorithms. • Example: BAG (Sun Kim) • 99 % accuracy (selective) but at cost of ~40-50 % fragmentation (over-sensitive) • Solution? • Bottom-Up merging back of fragmented clusters
Need for framework • Suggested bottom-up approach possible using various sub-methods • Framework: Do common and unique tasks seamlessly • Insert new sub-methods easily with very little hassle • Implemented primarily in Perl with supporting C programs and Unix Shell scripts
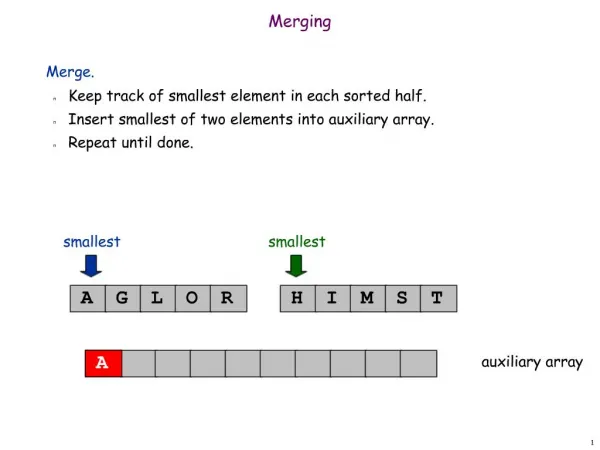
Framework Schematic Merge Suggestions from Clustering Algorithm Test Scaffold Generate Combined Profile for Two Fragment Clusters Prepare Sequence Data Post-process New Clustering Result Test Merge’bility Enhanced Clustering Result
Framework – Profile Generation Merge Suggestions from Clustering Algorithm Test Scaffold GENERATE COMBINED PROFILE FOR TWO FRAGMENT CLUSTERS Prepare Sequence Data Post-process New Clustering Result Test Merge’bility Enhanced Clustering Result
Profile Generation – MSA • MSA = Multiple Sequence Alignment C1 MSA (C1) Combined Profile MSA (C1, C2) C2 MSA (C2)
Profile Generation – MSA • Common first step: MSA profile generation for two fragment clusters C1 and C2 (Clustalw) • MSA (C1) and MSA (C2) • Most expensive step in framework • Common second step: Combined profile generation (Clustalw) • Prof_Align [MSA (C1), MSA (C2)]
Profile Generation – MSA explained.. • All of the implemented techniques depend on MSA profiles • MSA profile: align more than 2 sequences simultaneously Image from http://bioinformatics.weizmann.ac.il/~pietro/Making_and_using_protein_MA/
Profile Generation – MSA explained.. Image from http://www.mscs.mu.edu/~cstruble/class/mscs230/fall2002/notes/3
Framework – Merge’bility Test Merge Suggestions from Clustering Algorithm Test Scaffold Generate Combined Profile for Two Fragment Clusters Prepare Sequence Data Post-process New Clustering Result TEST MERGE’BILITY Enhanced Clustering Result
Model Comparison based Merge Test • Statistics/Machine learning technique based method: • Uses Relative Entropy and Statistical measures w.r.t. Runs test • Drawbacks • Almost impossible to nail down on threshold values for z-score or any other statistical measure • Extremely dependent sample size equality – does not work well when the two fragment sizes vary
Model Comparison based Merge Test • Each column in a MSA profile is a probabilistic model (details of construction beyond the scope of this talk) • Compute similarity between corresponding columns in the two fragments – Kullback Liebler distance • Need to consider gaps while matching up columns – challenging task • Also need to screen for random “good” distances – taken care off using random model in distance computation
Model Comparison based Merge Test • Using column wise comparison distance scores, compute “distance vector” • Symbolic representation for “good”, “bad” and “don’t care” distances (detail abstracted) • Do standard statistical test: Runs test to check out how random distance vector is… • Nice pattern: • y | y | y | n | n | y | y | y | n | n | y | y | y • Random pattern: • y | n | y | y | n | n | n | y | n | y | n | n | y | y
Model Comparison based Merge Test 4) Do Runs test
Model Comparison based Merge Test • Compute mean, standard deviation and subsequently z-score • Threshold to separate “good” and “bad” merges • Drawbacks again… • Threshold will be sample specific, hard to have one threshold for entire dataset (illustrated in test results) • Failure rate is high if sample size is unequal
Merge’bility Test – Techniques … • Phylogenetic tree based method: • Evolutionary Distance based method • Drawback: Too strict; many false negatives possible; Also hard to nail a threshold • Evolutionary Least Common Ancestor (LCA) based method • Improved performance in both of the previously mentioned issues
Phylogenetic Tree Distance based method • Clustalw (or other tree generation tools) provide NJ tree of a MSA profile • Sequence length normalized distance from root for each sequence • 0 < distance < 1 • Define some threshold for distance that constitutes intra/inter cluster distances
Phylogenetic Tree Distance based method • Distance between sequences from… • Two clusters will be closer to: • ‘1’ if two clusters are not merge’ble – call these “bad distances” • ‘0’ if two clusters are actually part of the same super cluster • The same cluster will be obviously closer to ‘0’ – these constitute “good distances”; don’t care in our case • Count number of “bad distances” • Gives a good idea of how good a merge is
Phylogenetic Tree Distance based method • Good enough? Not yet – need for normalization of the “bad distance” count. • Why? • Number of edges between vertices of same/different clusters is proportional to size of clusters!
Phylogenetic Tree Distance based method • Once normalization of number of “bad distances” is done, this method churned out decent results • Normalizing factor? Contentious.. What is a good normalizer? • Method too strict for unequally sized clusters. Most merges rejected leading to appreciable number of false negatives • Inherent nature of MSA programs and unequally sized profiles (cluster sizes)
Phy.Tree LCA coverage based method • Clustalw, Phylip (or other tree generation tools) provide a rooted phylogenetic tree for a MSA profile • Looking at the tree, one can easily make out if a pair of clusters should be merged or not • How? • Parse tree into a usual tree data structure and look for common ancestor of sequences of each cluster • Example…
Phy.Tree LCA coverage based method • Good Merge • Sequences of the two clusters (shaded blue and red) are from the same super cluster
Phy.Tree LCA coverage based method • Bad Merge • Sequences of the two clusters (shaded blue and red) are from different super clusters
Phy.Tree LCA coverage based method • Same LCA for both clusters? Good merge! • If not … Bad merge? • Not quite. Possible that LCAs may be different but they cover sequences from either cluster upto a considerable extent • Better to use coverage of LCAs instead • Example…
Phy.Tree LCA coverage based method • Why LCA Coverage? • Second cluster has three sequences, but its LCA covers four more sequences from the other cluster
Phy.Tree LCA coverage based method • Coverage test: • For clusters Ci and Ck, choose smaller cluster say Ci i.e | Ci | < | Ck | • Define Cov (LCA[Ci]) as the number of sequences LCA Ci covers. • If Cov(LCA[Ci]) > # of sequences in Ci … where | Ci | < | Ck | • i.e. { Cov (LCA[Ci]) / | Ci | } > 1 • Or {Cross Coverage (LCA[Ci])} > 0
Phy.Tree LCA coverage based method • Advantages: • Sample size difference does not play a big role • Demarcating between “good” and “bad” merges is much simpler and straight forward • Shown to work really well on a variety of data sizes, difficulty levels – test results… • Possible weakness: • Bound to fail for extremely small fragments (say 2 sequences each) – hard not to have a common LCA !
Acknowledgements! • A big thank you to: • Prof. Sun Kim, advisor • My parents, brother, grand parents! • All my colleagues and friends: JH, Zhiping, Scott Martin, SR, Raj, Anshul, Pat Hayes and everyone else! • Folks at CS & Informatics: CS Systems staff, Lucy, Linda, Wendy, Cheryl, Errissa, Bob! • Profs. Marty Siegel and Gary Wiggins – GPC. • RATS folks! • Did I forget someone?! Sorry if I did…