Download

1 / 20
200 likes | 336 Views
Protein threading algorithms. Presented by Jian Qiu. GenTHREADER Jones, D. T. JMB(1999) 287, 797-815 Protein Fold Recognition by Prediction-based Threading Rost, B., Schneider, R. & Sander, C. JMB(1997)270,471-480. Why do we need protein threading?.

E N D
Protein threading algorithms Presented by Jian Qiu • GenTHREADER Jones, D. T. JMB(1999) 287, 797-815 • Protein Fold Recognition by Prediction-based Threading • Rost, B., Schneider, R. & Sander, C. JMB(1997)270,471-480
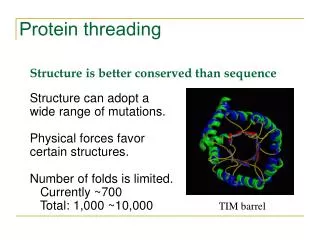
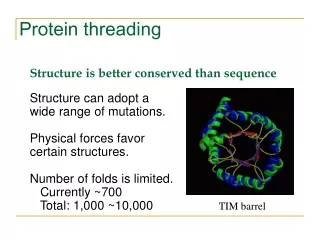
Why do we need protein threading? • To detect remote homologue Genome annotation Structures are better conserved than sequences. Remote homologues with low sequence similarity may share significant structure similarity. • To predict protein structure based on structure template Protein A shares structure similarity with protein B. We could model the structure of protein A using the structure of protein B as a starting point.
An successful example by GenTHREADER • ORF MG276 from Mycoplasma genitalium was predicted to share structure similarity with 1HGX. • MG276 shares a low sequence similarity (10% sequence identity) with 1HGX. • Supporting Evidence: • MG276 has an annotation of adeninephosphoribosyltransferase, basedon high sequencesimilarity tothe Escherichia coli protein; 1HGX isahypoxanthine-guanine-xanthinephosphoribosyltransferase from the protozoan parasite Tritrichomonas foetus. • Four functionally important residues in 1HGX are conserved in MG276. • The secondary structureprediction for ORFMG276 agrees very well with the observed secondary structure of 1HGX.
GenTHREADER Protocol Sequence alignment • For each template structure inthefold library, related sequences were collectedbyusingthe program BLASTP. • A multiple sequence alignment of these sequences was generated with a simplified version of MULTAL. • Get the optimal alignment between the target sequence and the sequence profile of a template structure with dynamic programming.
Threading Potentials Pairwise potential (the pairwise model family): k: sequence separation s: distance interval mab: number of pairs ab observedwithsequence separation k s: weight given toeachobservation fk(s): frequency of occurrence ofallresidue pairs fkab(s): frequency of occurrenceofresidue pair ab
Solvation potential (the profile model family): r: the degree of residue burial thenumber of other Cbatoms located within 10 Å of the residue'sCbatom fa(r): frequency of occurrence of residue awithburial r f (r): frequency ofoccurrenceof all residues with burial r
Variables considered to predict the relationship • Pairwise energy score • Solvation energy score • Sequence alignment score • Sequence alignment length • Length of the structure • Length of the target sequence
Artificial Neural Network A node
The effects of sequence alignment score and pairwise potential on the Network output
Confidence level with different network scores Medium(80%) High (99%) Certain (100%) Low
Genome analysis of Mycoplasma genitalium All the 468 ORFs were analyzed within one day.
PHD: Predict 1D structure from sequence Sequence MaxHom Multiple Sequence Alignment PHDsec PHDacc Secondary structure: H(helix), E(strand), L(rest) Solvent accessibility: Buried(<15%), Exposed(>=15%)
Similarity matrix in dynamic programming • Purely structure similarity matrix: six states (combination of three secondary structure states and two solvent accessibility states) • Purely sequence similarity matrix: McLachlan or Blosum62 • Combination of strcture and sequence similarity matrix: Mij=m*Mij1D structure + (100-m)*Mijsequence m=0: sequence alignment only m=100: 1Dstructure alignment only
Results on the 11 targets of CASP1 • Correctly detected the remote homologues at first rank in four cases; Average percentage of correctly aligned residues: 21%; Average shift: nine residues. Best performing methods in CASP1: • Expert-driven usage of THREADER by David Jones and colleagues detected five out of nine proteins correctly at first rank. • Best alignments of the potential-based threading method by Manfred Sippl and colleagues were clearly better than the best ones of this algorithm.