Download
1 / 1
10 likes | 137 Views
Limited Proteolysis of MDH with Immobilized TPCK-Trypsin.

E N D
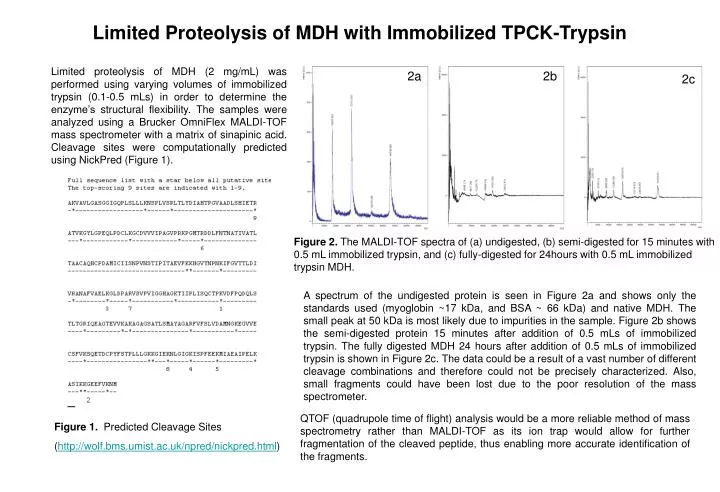
Limited Proteolysis of MDH with Immobilized TPCK-Trypsin Limited proteolysis of MDH (2 mg/mL) was performed using varying volumes of immobilized trypsin (0.1-0.5 mLs) in order to determine the enzyme’s structural flexibility. The samples were analyzed using a Brucker OmniFlex MALDI-TOF mass spectrometer with a matrix of sinapinic acid. Cleavage sites were computationally predicted using NickPred (Figure 1). 2a 2b 2c Figure 2. The MALDI-TOF spectra of (a) undigested, (b) semi-digested for 15 minutes with 0.5 mL immobilized trypsin, and (c) fully-digested for 24hours with 0.5 mL immobilized trypsin MDH. A spectrum of the undigested protein is seen in Figure 2a and shows only the standards used (myoglobin ~17 kDa, and BSA ~ 66 kDa) and native MDH. The small peak at 50 kDa is most likely due to impurities in the sample. Figure 2b shows the semi-digested protein 15 minutes after addition of 0.5 mLs of immobilized trypsin. The fully digested MDH 24 hours after addition of 0.5 mLs of immobilized trypsin is shown in Figure 2c. The data could be a result of a vast number of different cleavage combinations and therefore could not be precisely characterized. Also, small fragments could have been lost due to the poor resolution of the mass spectrometer. QTOF (quadrupole time of flight) analysis would be a more reliable method of mass spectrometry rather than MALDI-TOF as its ion trap would allow for further fragmentation of the cleaved peptide, thus enabling more accurate identification of the fragments. Figure 1. Predicted Cleavage Sites (http://wolf.bms.umist.ac.uk/npred/nickpred.html)