Download

1 / 24
240 likes | 337 Views
Bayesian Refinement of Protein Functional Site Matching. Kanti V Mardia , Vysaul B Nyirongo *, Peter J Green, Nicola D Gold, David R Westhead. Presented by Deephan , Mohan. Presentation Flow.

E N D
Bayesian Refinement of Protein Functional Site Matching Kanti V Mardia, Vysaul B Nyirongo*, Peter J Green, Nicola D Gold, David R Westhead Presented by Deephan, Mohan
Presentation Flow Disclaimer : Contrary to the assumption made by the authors, the paper presenter does have a thorough understanding of all the concepts related to the topics of advanced statistical, graph theory and structural genomics discussed in the paper.. Background Conventional Methods Bayesian Refinement Results Conclusion
Motivation • Structural Genomics • Structural Site comparison • Functional Site comparison • Knowledge based methods • Similarity Search Algorithms
Protein Functional Site Matching • Modeled as a graph theoretic problem • Shape analysis of Proteins • Crucial for prediction of molecular interactions • Infer functional relationship of proteins • Classification of Binding Patterns • Resource: SITESDB Database • Contains Protein Structural data • Entries formed from PDB (Protein Data Bank)
The Methodology • Graph Similarity Problem • Objective: Matching Functional sites -Comparing amino acid configurations (Cα and Cβ atoms) • Functional site – Graph • Amino acid positions – Vertices • Refining the Graph Match • Application of Bayesian Strategy • Markov Chain Monte Carlo (MCMC) procedure
Need for Bayesian Refinement?? Bayesian Inference: • Complete Distribution of matches • Solution space • Noise Adaptation • Flexibility • Edge over combinatorial methods
Bayesian Model • Common Tool used in Statistical Inference • Based on Posterior Joint Distribution • Product of Prior density and Likelihood Biologically speaking, • Prior Density - Distribution of Transformation Parameters • Likelihood - Related to matches between functional sites
Representation and Matching Functional sites X and Y represented as Graphs G1 and G2 Vertex sets V1 = {Xj, j = 1, 2, ..., m} , V2 = {Yk, k = 1, 2, ..., n} Xj , Yk - represents coordinates of amino acids in jth and kth positions of X,Y x1j, y1k – Cα coordinates for X,Y x2j, y2k – Cβ coordinates for X,Y x1 = {x1j : j = 1 ..., m}, x2 = {x2j : j = 1 ..., m} y1 = {y1k : k = 1 ..., n}, y2 = {y2k : k = 1 ..., n}
Graph Theoretic Approach • Objective: • Creation of Vertex Product Graph (Hv) • Hv = G1 ○v G2 • VH=V1 x V2 • An edge between two vertices vh = (Xj, Yk), vh' = (Xj', Yk') ∈ VH exists for j ≠ j' and k ≠ k' when • 1. the absolute difference between distances |x1j - x1j'| and |y1k - y1k'| and • 2. also the absolute difference between distances |x2j - x2j'| and | y2k - y2k'| are both less than 1.5Å (matching distance threshold).
Bayesian Alignment 1 if jth amino acid corresponds to kth amino acid 0 otherwise • Matching between amino acids X and Y represented by matrix M, Mjk = • Transformations to bring the configurations into alignment is given by • xij = Ayik + τ for Mjk = 1, i = 1, 2 A – Rotation Matrix, τ – Translation vector
Bayesian Modeling (contd) Joint Posterior Distribution: p(A), p(τ) and p(σ) denote prior distributions for A, τ and σ |A| - Jacobian Transformation presence of Gaussian noise N(0, σ2) in in the atomic positions for x1j and y1k
Bayesian Modeling (contd) Side chains orientation: Extending the model by taking into account the relative orientation of Cα and Cβin matching amino acids
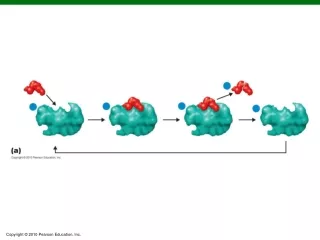
MCMC Refinement Step Markov Chain Monte Carlo (MCMC) – used to sample the full joint distribution function p(M, A, τ, σ, x1, y1, x2, y2) p(M, A, τ, σ, x1, y1, x2, y2) – function of RMSD and anglefororientationdifferencebetween amino acids
Significance of RMSD RMSD – Root Mean Square Distribution Matches of lower RMSD over larger numbers of matching residues are more statistically significant MCMC Refinement improved the RMSD (reduction) and the number of matching residues ( increase)
Decision tree for refining the graph solution by the MCMC method. Boxes with curved corners show processes and their output while boxes with sharp corners are for branching conditions. The procedure starts with graph solution MG. The graph solution's RMSD and number of matches are denoted by RMSDG and LG respectively. MCMC is re-iterated until the MCMC solution: MB is better. The RMSD and number of matches for MB are denoted by RMSDB and LB respectively. MB and MG are compared using 1) RMSDs and the number of matches or 2) P-values for MG and MG, denoted by PG and PB respectively.
Results • Two Binding Sites: • Alcohol dehydrogenase structure (60 amino acids) • 17 – β hydroxysteroiddehydrogenase ( 63 amino acids) • 4 Matching Studies were performed • Each study was performed with and without considering the physico-chemical properties of amino-acids.
Case 1: Site 1hdx_1 matching against its own SCOP family • 125/145 sites produced significant matches – increased to 131/145 (after refinement) • RMSD is improved • from > 1.5Å to less than • 1Å • Increase in the number of matching residues Case-I
Case 2: 17 – β hydroxysteroiddehydrogenase and family • After MCMC Refinement step significant matches increased from 248 to 318 of 326 sites • Increased number of matching residues at a similar RMSD • RMSD improvement in minority of the sites
Case 3: alcohol dehydrogenase and superfamily • Matching sites increased form 200 to 324 • Case 4: Alcohol dehydrogenase and FAD/NAD(P)-binding domain • 12 sites improved after MCMC refinement
Discussion of Results MCMC refinement step provides significant improvement over Graph Matching Techniques Success – Lack of dependence on strict distance matching criteria Computationally expensive Refinements adapts to shape variations in binding sites