Download

1 / 29
551 likes | 2.05k Views
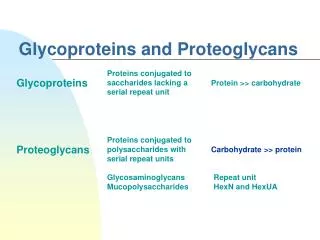
GLYCOPROTEINS. ASSOC. PROF. DR. CEMİLE KOCA ANKARA ATATÜRK TRAINING AND RESEARCH HOSPITAL. Glycoproteins and Proteoglycans. Proteins conjugated to polysaccharides with serial d isac. repeat units. Carbohydrate >> protein. Proteoglycans. Glycosaminoglycans Mucopolysaccharides.

E N D
GLYCOPROTEINS ASSOC. PROF. DR.CEMİLE KOCA ANKARA ATATÜRK TRAINING AND RESEARCH HOSPITAL
Glycoproteins and Proteoglycans Proteins conjugated to polysaccharides with serial disac. repeat units Carbohydrate >> protein Proteoglycans Glycosaminoglycans Mucopolysaccharides Repeat unit HexN and HexUA Proteins conjugated to oligosacch.lacking a serial repeat unit may or may not be negatively charged Protein >> carbohydrate CHO chain is relatively short usually 2-10 sugar residues in length, but generally branched CHOs, primarilyD-hexoses Glycoproteins
Outer face of plasma membrane, ECM, blood • Inside cells; golgi, secretory granules, lysosomes
Types of glycoproteins • Membrane proteins • Secreted proteins • Antibody • hormones • milk proteins (lactalbumin) • proteins released by the pancreas • lysosomal proteins
Functions of Glycoproteins • Membrane-bound glycoproteins • Specific sites for recognition by other cells, hormones, viruses… (information-rich oligosaccharide) • cell surface antigenicity (such as the blood group antigens)
Blood ABO Antigens Functions of Glycoproteins O-linked oligosaccharides on the surface of RBCs help provide the ABO blood group determinants open square = GlcNAc open diamond = galactose filled square = fucose filled diamond = GalNAc filled diamond = sialic acid (NANA)
Functions of Glycoproteins • - components of the ECM • - components of the mucinsof the gastrointestinal & urogenital tracts, where they act as protective biologic lubricants • - almost all of the globular proteinspresent in human plasma are glycoproteins
Three main types according to glycosylation to proteins: 1. N-linked glycosylation : Asn N-glycans predominates in serumglycoproteins 2. O-linkedglycosylation : Ser, Thr membrane bound or ECM glycoprot, mucins 3. GPI(glycosyl phosphatidylinositol) anchored saccaharide core consisting of Man3GlcNH2, a bridge between the phospholipid in the membrane and the protein
Three Main Types of Glycoprotein Structures GPI-linked O-linked GPI = Glycosylphosphatidylinositol N-linked
N-linked oligosaccharides: • 2 broad classes: • Complexoligosacch’s • High-mannose oligosacch’s. • both contain same core pentasaccharide • but the complex oligosacch’s contain a diverse group of additional sugars, • e.g., N-acetylglucosamine (GlcNAc), L-fucose (Fuc), N-acetylneuraminic acid (NANA), • whereas the high-mannose oligosacch’s contain primarily mannose (Man) Complex oligosacc high-mannose oligosacc.
Glycoproteins • Advantages of oligosaccharide attachment • Hydrophilicity (polarity & solubility) • Destination label • Label for protein quality control • Protein folding • Protection from proteolytic attacks • Informational roles (recognition & communication) • targeting signal for removal of damaged or mis-folded proteins from the cell A glycoprotein may contain only one type of glycosidic linkage (N- or O-linked), or may have both O- and N-linked oligosacchs within same molecule
Synthesis of glycoproteins • Cytoplasmic proteins are synthesized on free ribosomes in cytosol • proteins, including many glycoproteins, that are destined for cellular memb’s, lysosome, or to be exported from cell, are synthesized on ribosomes attached to rER. • These proteins contain specific signal sequences at their N-terminal end that act as molecular “address labels” which direct the proteins to their proper destinations Transport of glycoproteins through the Golgi apparatus and their subsequent release
Synthesis of Carbohydrate components of glycoproteins • Glycosyltransferaseenzymes located on membranes of the endoplasmicreticulum and Golgi apparatus • The precursors of the CHO components of glycoproteins are sugar nucleotides • which include UDP-glucose, UDP-galactose, UDP-N-acetylglucosamine & UDP-N-acetylgalactosamine • In addition, GDP-mannose, GDP-L-fucose (which is synthesized from GDP-mannose), & CMP-N-acetylneuraminic acid may donate sugars to the growing chain
Synthesis of O-linked glycosides • O-linked glycoproteins are synthesized by the sequential transfer of sugars from their nucleotide carriers to the protein. • 1st the protein to which the oligosacch’s are to be attached is synthesized on the rER, & extruded into its lumen • Posttranslational Glycosylation • begins with the transfer of an N-acetylgalactosamine (from UDP-N-acetylgalactosamin) onto a specific seryl or threonyl R-group
also occurs in the lumen of the ER & in Golgi Cotranslational glycosylation The protein itself does not become glycosylated with individual sugars transfer of a pre-formed oligosacch from its carrier, a membrane lipid(dolichol)& its phosphorylated derivative, dolichol pyrophosphateto the protein Synthesis of the N-linked glycosides
The Golgi Apparatus has two major functions: • Modifies the N-linked • oligosaccharides and adds O-linked oligosaccharides • 2. Sorts proteins so that when they exit the trans Golgi network, they are delivered to the correct destination
Roles of Oligosaccharide Recognition and Adhesion at the Cell Surface
Targeting of Lysosomal Enzymes to Lysosomes The phosphate is added in the Golgi N-linked glycoproteins being processed through Golgi can be phosphorylated at one or or more specific mannosyl residues This was first attached in the ER. Addition of mannose 6-phosphateM6P as chemical marker to lysosomal enzymes
Targeting of Lysosomal Enzymes to Lysosomes Mannose 6-P receptors, located in Golgi, bind the mannose 6-P residues of these targeted enz’s, resulting in their translocation to the lysosomes Acidic pH causes hydrolase to dissociate from the receptor.
Lysosomal degradation of glycoproteins • Glycoproteins are degraded in lysosomes by acid hydrolases similar to that of GAGs. • “last on, first off” • A deficiency of one of these enz’s results in a glycoprotein storage disease (oligosaccharidosis), resulting in accumulation of partially degraded structures in the lysosome • After cell death, the oligosacch fragments appear in the urine • Disorders are very often directly associated with the same enzyme deficiencies involved in mucopolysaccharidoses & the inability to degrade glycolipids
Mechanism for transport of N-linked glycoproteins to the lysosomes
Inclusion-cell disease (I-CELL DISEASE) • a rare syndromein which the acid hydrolase enz’s normally found in lysosomes are absent, resulting in an accumulation of substrates normally degraded by lysosomal enz’s within these vesicles • individuals with I-cell disease are lacking the enzymic ability to phosphorylate the mannose residues of potential lysosomal enz’s • causing an incorrect targeting of these proteins to extracellular sites (high amounts in plasma), rather than lysosomal vesicles • I-cell disease is so-named because of the large inclusion bodies seen in cells of patients with this disease
I-cell disease is characterized by; • skeletal abnormalities, • restricted joint movement, • coarse facial features • severe psychomotor impairment • Death (usually by age 8 yrs) I-CELL DISEASE