Download
1 / 51
510 likes | 609 Views
Signaling Mechanisms, Cellular Adhesion, and More diseases. Genetics. Signal Transduction. Occurs on the cell membrane on both side Cells n a multicellular organism need to communicate with other cells

E N D
Signaling Mechanisms, Cellular Adhesion, and More diseases Genetics
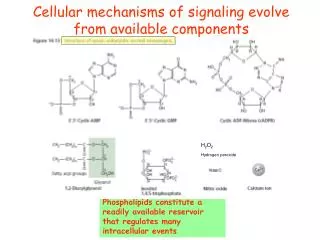
Signal Transduction • Occurs on the cell membrane on both side • Cells n a multicellular organism need to communicate with other cells • In signal transduction molecules on the cell membane, assist, transmit,and amplify messages. • Transduction means to change the signal from the environment into a common message understood by the cell.
Signal transduction • The transduction occurs between a receptor on the outside of the cell and a molecule in the cytoplasm which will amplify the signal so that it is received and acted on
First messengers • Messages received by receptors on the outside • These are the first messengers – the signal can be light, a chemical gradient,temperature, toxins, hormones, or growth factors
Receptor • A molecule which is the signal binds to the receptor. • The receptor is attached to a transmembrane protein( spans the cell membrane) • The signal causes a change in the shape or conformation of the transmembrane protein
Cytoplasmic responses • The membrane affects a regulator molecule that then activates an enzyme • The enzyme causes ATP to form CAMP( cyclic AMP) • This is a generalized message that can be transmitted to the nucleus or to other molecules in the cell
NF1 and Growth factors • Chromosome 17 • Neurofibromatosis is an autosomal dominant disorder characterized particularly by cafe-au-lait spots and fibromatous tumors of the skin. Other features are variably present • Caused by a mutation in the gene for neurofibromin
NF1 and cancer • Neurofibromatosis is an autosomal dominant disorder characterized particularly by cafe-au-lait spots and fibromatous tumors of the skin. Other features are variably present
Protein • The protein has been called neurofibromin 1; 2839 amino acids • Expression is tissue and development stage specific • Function GTPase activating protein • (GAP) interacting with p21RAS -> tumor suppressor.
Cellular Adhesion • Cells touch each other through adhesion • A precise set of interactions between proteins joins the cells in tissues
Cellular Adhesion • Inflammation – the painful,red swelling at a site of injury or infection- illustrates cell adhesion • Inflammation is caused by white blood cells. White blood cells flood to injured areas to prevent infection • Cellular adhesion molecules help guide white blood cells to the injured area( genetically controlled)
Three Types of CAMS – cellular adhesion molecules • Selectins provide traction by coating the white blood cells to slow them • Blood cells release chemical attractants that signal white blood cells to stop this activatesCAMs called integrins that latch onto white blood cells and CAMs called adhesions receptor proteins
Adhesion receptor • This extends from the capillary wall at the injury site and touches the cytoskeleton beneath the capillary lining • The integin and receptor protein bind the WBC and pull in through the membrane to the injury site
Failure to work • Creates a disease • Called leukocyte adhesion deficiency • A deficiency of CAMS • The blood does not stop at injured places • Lack of cell adhesion allows cells to travel through the body and metasticize
Deficiency of CAMs • Can also lead to arthritic situations where WBC attach to a joint when there is not injury
LAD • LAD is a rare PI disease, found in one out of every million people. This disease causes recurrent, life-threatening infections. Phagocytes cannot find their way to the site of infection to fight off invading germs. LAD is autosomal recessive disease, meaning that to be born with this disease, both parents must have the affected gene.
Cause • LAD is caused by a lack of beta 2 integrin, also called CD18, molecules. These molecules are normally found on the outer surface of phagocytes. Without them, the phagocytes cannot attach to blood vessel walls and enter infected tissues where they help fight infection. Mutations in the gene that instructs, or codes for, the production of CD18 cause LAD.
Infections and LAD • Children with LAD cannot fight off infection properly. They may have • Severe infections of the soft tissue • Eroding skin sores without pus • Severe infections of the gums with tooth loss • Infections of the gastrointestinal tract • Wounds that heal slowly and may leave scars
Lesch- Nyhan • Lesch-Nyhan syndrome (LNS) is a rare, inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT). LNS is an X-linked recessive disease-- the gene is carried by the mother and passed on to her son. LNS is present at birth in baby boys.
HPRT • The lack of HPRT causes a build-up of uric acid in all body fluids, and leads to symptoms such as severe gout, poor muscle control, and moderate retardation, which appear in the first year of life. A striking feature of LNS is self-mutilating behaviors – characterized by lip and finger biting – that begin in the second year of life.
Uric acid • Abnormally high uric acid levels can cause sodium urate crystals to form in the joints, kidneys, central nervous system, and other tissues of the body, leading to gout-like swelling in the joints and severe kidney problems.
Symptoms • Neurological symptoms include facial grimacing, involuntary writhing, and repetitive movements of the arms and legs similar to those seen in Huntington’s disease. Because a lack of HPRT causes the body to poorly utilize vitamin B12, some boys may develop a rare disorder called megaloblastic anemia.
MSUD – Maple Syrup Urine Disease • Maple Syrup Urine Disease (MSUD) or branched-chain ketoaciduria is caused by a deficiency in activity of the branched-chain a-ketoacid dehydrogenase (BCKD) complex (1, 2). This metabolic block results in the accumulation of the branched-chain amino acids (BCAAs) leucine, isoleucine, and valine and the corresponding branched-chain alpha-keto acids (BCKAs).
Characteristics • MSUD is an autosomal recessive metabolic disorder of panethnic distribution. The worldwide frequency based on routine screening data from 26.8 million newborns is approximately one in 185,000. In the inbred Old Order Mennonite population of Lancaster and Lebanon Counties, Pennsylvanis, MSUD occurs in approximately one in 176 newborns.
Protein • The human BCKD complex affected in MSUD is a macromolecular metabolic machine (molecular mass 4 x 106 daltons) loosely associated with the inner membrane of the mitochondria.
MSUD- Synthetic Diet • The diet centers around a synthetic formula or "medical food" which provides nutrients and all the amino acids except leucine, isoleucine and valine. These three amino acids are added to the diet with carefully controlled amounts of food to provide the protein necessary for normal growth and development without exceeding the level of tolerance.
Wilson’s Disease • Wilson's disease causes the body to retain copper. The liver of a person who has Wilson's disease does not release copper into bile as it should.
Symptoms • Wilson's disease is hereditary. Symptoms usually appear between the ages of 6 and 20 years, but can begin as late as age 40. The most characteristic sign is the Kayser-Fleischer ring—a rusty brown ring around the cornea of the eye that can be seen only through an eye exam.
Liver and Spleen • Other signs depend on whether the damage occurs in the liver, blood, central nervous system, urinary system, or musculoskeletal system. Many signs can be detected only by a doctor, like swelling of the liver and spleen
Other symptoms • Some symptoms are more obvious, like jaundice, which appears as yellowing of the eyes and skin; vomiting blood; speech and language problems; tremors in the arms and hands; and rigid muscles.
Treatment • The disease is treated with lifelong use of D-penicillamine or trientine hydrochloride, drugs that help remove copper from tissue, or zinc acetate, which stops the intestines from absorbing copper and promotes copper excretion
Dietary Restrictions • Patients will also need to take vitamin B6 and follow a low-copper diet, which means avoiding mushrooms, nuts, chocolate, dried fruit, liver, and shellfish.
Epidermolysis bullosa • Epidermolysis bullosa (EB) is a group of inherited bullous disorders characterized by blister formation in response to mechanical trauma. Historically, EB subtypes have been classified according to skin morphology.
Types • EB is classified into 3 major categories, including (1) EB simplex (EBS; intraepidermal skin separation), (2) junctional EB (JEB; skin separation in lamina lucida or central BMZ), and (3) dystrophic EB (DEB; sublamina densa BMZ separation; see
Biotinidase deficiency • Biotinidase is a ubiquitous mammalian cell enzyme occurring at high levels in the liver, serum, and kidney. The primary function is to cleave biotin from biocytin, preserving the pool of biotin for use as a cofactor for biotin dependent enzymes, namely the 4 human carboxylases
Disease caused by complete or partial absence of the enzyme is associated with a wide spectrum of clinical manifestations, including abnormalities of the neurological, dermatological, immunological, and ophthalmological systems. In spite of its rarity, early recognition is crucial because expeditious treatment may reverse all of its manifestations
Treatment • If treated promptly, biotinidase deficiency may be asymptomatic. Prolonged symptoms prior to institution of biotin therapy may leave the patient with varying degrees of neurological sequelae, including mental retardation, seizures, and coma. Death may result from untreated profound biotinidase deficiency.
Neurological Problems • Developmental delay • Ataxia • Neuropathy • Auditory nerve dysfunction