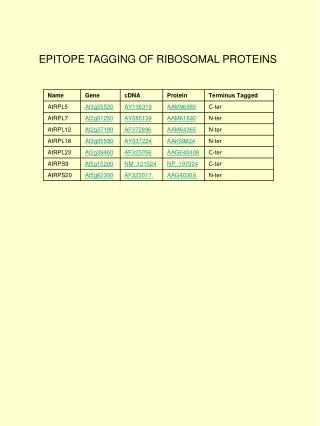
Download

1 / 36
410 likes | 665 Views
Epitope prediction algorithms. Urmila Kulkarni-Kale Bioinformatics Centre University of Pune. Vaccine development In Post-genomic era: Reverse Vaccinology Approach. Rappuoli R. (2000). Reverse vaccinology. Curr Opin Microbiol. 3:445-450. . Genome Sequence. Proteomics Technologies.

E N D
Epitope prediction algorithms Urmila Kulkarni-Kale Bioinformatics Centre University of Pune
Vaccine development In Post-genomic era: Reverse Vaccinology Approach. • Rappuoli R. (2000). Reverse vaccinology. Curr Opin Microbiol. 3:445-450. © Bioinformatics Centre, UoP
Genome Sequence Proteomics Technologies In silico analysis DNA microarrays High throughput Cloning and expression In vitro and in vivo assays for Vaccine candidate identification Global genomic approach to identify new vaccine candidates © Bioinformatics Centre, UoP
In Silico Analysis Peptide Multiepitope vaccines VACCINOME Candidate Epitope DB Epitope prediction Disease related protein DB Gene/Protein Sequence Database © Bioinformatics Centre, UoP
Types of Epitopes • Sequential / Continuous epitopes: • recognized by Th cells • linear peptide fragments • amphipathic helical 9-12 mer • Conformational / Discontinuous epitopes: • recognized by both Th & B cells • non-linear discrete amino acid sequences, come together due to folding • exposed 15-22 mer © Bioinformatics Centre, UoP
Properties of Epitopes • They occur on the surface of the protein and are more flexible than the rest of the protein. • They have high degree of exposure to the solvent. • The amino acids making the epitope are usually charged and hydrophilic. © Bioinformatics Centre, UoP
Methods to identify epitopes • Immunochemical methods • ELISA : Enzyme linked immunosorbent assay • Immunoflurorescence • Radioimmunoassay • X-ray crystallography: Ag-Ab complex is crystallized and the structure is scanned for contact residues between Ag and Ab. The contact residues on the Ag are considered as the epitope. • Prediction methods: Based on the X-ray crystal data available for Ag-Ab complexes, the propensity of an amino acid to lie in an epitope is calculated. © Bioinformatics Centre, UoP
Antigen-Antibody (Ag-Ab) complexes • Non-obligatory heterocomplexes that are made and broken according to the environment • Involve proteins (Ag & Ab) that must also exist independently • Remarkable feature: • high affinity and strict specificity of antibodies for their antigens. • Ab recognize the unique conformations and spatial locations on the surface of Ag • Epitopes & paratopesare relational entities © Bioinformatics Centre, UoP
Antigen-Antibody complex © Bioinformatics Centre, UoP
Sequential Conformational Ab-binding sites Ab-binding sites:Sequential & Conformational Epitopes! Paratope © Bioinformatics Centre, UoP
B cell epitope prediction algorithms : • Hopp and Woods –1981 • Welling et al –1985 • Parker & Hodges -1986 • Kolaskar & Tongaonkar – 1990 • Kolaskar & Urmila Kulkarni - 1999 T cell epitope prediction algorithms : • Margalit, Spouge et al - 1987 • Rothbard & Taylor – 1988 • Stille et al –1987 • Tepitope -1999 © Bioinformatics Centre, UoP
Hopp & Woods method • Pioneering work • Based on the fact that only the hydrophilic nature of amino acids is essential for an sequence to be an antigenic determinant • Local hydrophilicity values are assigned to each amino acid by the method of repetitive averaging using a window of six • Not very accurate © Bioinformatics Centre, UoP
Welling’s method • Based on the % of each aa present in known epitopes compared with the % of aa in the avg. composition of a protein. • assigns an antigenicity value for each amino acid from the relative occurrence of the amino acid in an antigenic determinant site. • regions of 7 aa with relatively high antigenicity are extended to 11-13 aa depending on the antigenicity values of neighboring residues. © Bioinformatics Centre, UoP
Parker & Hodges method • Utilizes 3 parameters : • Hydrophilicity : HPLC • Accessibility : Janin’s scale • Flexibility : Karplus & Schultz • Hydrophilicity parameter was calculated using HPLC from retention co-efficients of model synthetic peptides. • Surface profile was determined by summing the parameters for each residue of a seven-residue segment and assigning the sum to the fourth residue. • One of the most useful prediction algorithms © Bioinformatics Centre, UoP
Kolaskar & Tongaonkar’s method • Semi-empirical method which uses physiological properties of amino acid residues • frequencies of occurrence of amino acids in experimentally known epitopes. • Data of 169 epitopes from 34 different proteins was collected of which 156 which have less than 20 aa per determinant were used. • Antigen: EMBOSS © Bioinformatics Centre, UoP
CEP Server • Predicts the conformational epitopes from X-ray crystals of Ag-Ab complexes. • uses percent accessible surface area and distance as criteria © Bioinformatics Centre, UoP
An algorithm to map sequential and conformational epitopes of protein antigens of known structure © Bioinformatics Centre, UoP
CE: Beyond validation • High accuracy: • Limited data set to evaluate the algorithm • Non-availability of true negative data sets • Prediction of false positives? • Are they really false positives? • Limitation: • Limited by the availability of 3D structure data of antigens Different Abs (HyHEL10 & D1.3) have over-lapping binding sites © Bioinformatics Centre, UoP
CE: Features • The first algorithm for the prediction of conformational epitopes or antibody binding sites of protein antigens • Maps both: sequential & conformational epitopes • Prerequisite: 3D structure of an antigen © Bioinformatics Centre, UoP
CEP: Conformational Epitope Prediction Serverhttp://bioinfo.ernet.in/cep.htm © Bioinformatics Centre, UoP
T-cell epitope prediction algorithms • Considers amphipathic helix segments, tetramer and pentamer motifs (charged residues or glycine) followed by 2-3 hydrophobic residues and then a polar residue. • Sequence motifs of immunodominant secondary structure capable of binding to MHC with high affinity. • Virtual matrices which are used for predicting MHC polymorphism and anchor residues. © Bioinformatics Centre, UoP
Case study: Design & development of peptide vaccine against Japanese encephalitis virus © Bioinformatics Centre, UoP
We Have Chosen JE Virus, Because • JE virus is endemic in South-east Asia including India. • JE virus causes encephalitis in children between 5-15 years of age with fatality rates between 21-44%. • Man is a "DEAD END" host. © Bioinformatics Centre, UoP
We Have Chosen JE Virus, Because • Killed virus vaccine purified from mouse brain is used presently which requires storage at specific temperatures and hence not cost effective in tropical countries. • Protective prophylactic immunity is induced only after administration of 2-3 doses. • Cost of vaccination, storage and transportation is high. © Bioinformatics Centre, UoP
Predicted structure of JEVSMutations: JEVN/JEVS © Bioinformatics Centre, UoP
CE of JEVN Egp © Bioinformatics Centre, UoP
Species and Strain specific properties:TBEV/ JEVN/JEVS • Loop1 in TBEV: LA EEH QGGT • Loop1 in JEVN: HN EKR ADSS • Loop1 in JEVS: HN KKR ADSS Antibodies recognising TBEV and JEVN would require exactly opposite pattern of charges in their CDR regions. Further, modification in CDR is required to recognise strain-specific region of JEVS. © Bioinformatics Centre, UoP
Multiple alignment of Predicted TH-cell epitope in the JE_Egp with corresponding epitopes in Egps of other Flaviviruses • 426457 • JE DFGSIGGVFNSIGKAVHQVFGGAFRTLFGGMS • MVE DFGSVGGVFNSIGKAVHQVFGGAFRTLFGGMS • WNE DFGSVGGVFTSVGKAIHQVFGGAFRSLFGGMS • KUN DFGSVGGVFTSVGKAVHQVFGGAFRSLFGGMS • SLEDFGSIGGVFNSIGKAVHQVFGGAFRTLFGGMS • DEN2DFGSLGGVFTSIGKALHQVFGAIYGAAFSGVS • YFDFSSAGGFFTSVGKGIHTVFGSAFQGLFGGLN • TBEDFGSAGGFLSSIGKAVHTVLGGAFNSIFGGVG • COMM DF S GG S GK H V G F G • Multiple alignment of JE_Egp with Egps of other Flaviviruses in the YSAQVGASQ region. • 151 183 • JE SENHGNYSAQVGASQAAKFTITPNAPSITLKLG • MVE STSHGNYSTQIGANQAVRFTISPNAPAITAKMG • WNE VESHG‑‑‑‑KIGATQAGRFSITPSAPSYTLKLG • KUN VESHGNYFTQTGAAQAGRFSITPAAPSYTLKLG • SLE STSHGNYSEQIGKNQAARFTISPQAPSFTANMG • DEN2 HAVGNDTG‑‑‑‑‑KHGKEIKITPQSSTTEAELT • YF QENWN‑‑‑‑‑‑‑‑TDIKTLKFDALSGSQEVEFI • TBE VAANETHS‑‑‑‑GRKTASFTIS‑‑SEKTILTMG © Bioinformatics Centre, UoP
Peptide Modeling • Initial random conformation • Force field: Amber • Distance dependent dielectric constant 4rij • Geometry optimization: Steepest descents & Conjugate gradients • Molecular dynamics at 400 K for 1ns • Peptides are: • SENHGNYSAQVGASQ • NHGNYSAQVGASQ • YSAQVGASQ • YSAQVGASQAAKFT • NHGNYSAQVGASQAAKFT • SENHGNYSAQVGASQAAKFT • 149 168
Relevant Publications & Patent • Urmila Kulkarni-Kale, Shriram Bhosale, G. Sunitha Manjari, Ashok Kolaskar, (2004). VirGen: A comprehensive viral genome resource. Nucleic Acids Research 32:289-292. • Urmila Kulkarni-Kale & A. S. Kolaskar (2003). Prediction of 3D structure of envelope glycoprotein of Sri Lanka strain of Japanese encephalitis virus. In Yi-Ping Phoebe Chen (ed.), Conferences in research and practice in information technology. 19:87-96. • A. S. Kolaskar & Urmila Kulkarni-Kale (1999) Prediction of three-dimensional structure and mapping of conformational antigenic determinants of envelope glycoprotein of Japanese encephalitis virus. Virology. 261:31-42. Patent: Chimeric T helper-B cell peptide as a vaccine for Flaviviruses. Dr. M. M. Gore, Dr. S.S. Dewasthaly, Prof. A.S. Kolaskar, Urmila Kulkarni-Kale Sangeeta Sawant WO 02/053182 A1 © Bioinformatics Centre, UoP
Important references • Hopp, Woods, 1981, Prediction of protein antigenic determinants from amino acid sequences, PNAS U.S.A 78, 3824-3828 • Parker, Hodges et al, 1986, New hydrophilicity scale derived from high performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and X-ray derived accessible sites, Biochemistry:25, 5425-32 • Kolaskar, Tongaonkar, 1990, A semi empirical method for prediction of antigenic determinants on protein antigens, FEBS 276, 172-174 • Men‚ndez-Arias, L. & Rodriguez, R. (1990), A BASIC microcomputer program forprediction of B and T cell epitopes in proteins, CABIOS, 6, 101-105 • Peter S. Stern (1991), Predicting antigenic sites on proteins, TIBTECH, 9, 163-169 • A.S. Kolaskar and Urmila Kulkarni-Kale, 1999 - Prediction of three-dimensional structure and mapping of conformational epitopes of envelope glycoprotein of Japanese encephalitis virus,Virology, 261, 31-42 © Bioinformatics Centre, UoP