Download

1 / 36
380 likes | 593 Views
Genomes. Daniel Lawson VectorBase @ EBI. Bioinformatic Tools for Comparative Genomics of Vectors. Tuesday 10:30 - 13:00 Genome sequencing 14:00 - 16:00 Genome annotation 16:30 - 18:00 Practical Wednesday 9:30 - 10:00 Review genome annotation 10:30 - 13:00 Comparative genomics I

E N D
Genomes Daniel Lawson VectorBase @ EBI Bioinformatics Tools for Comparative Genomics of Vectors
Bioinformatic Tools for Comparative Genomics of Vectors • Tuesday • 10:30 - 13:00 Genome sequencing • 14:00 - 16:00 Genome annotation • 16:30 - 18:00 Practical • Wednesday • 9:30 - 10:00 Review genome annotation • 10:30 - 13:00 Comparative genomics I • 14:00 - 16:00 Comparative genomics II • 16:30 - 18:00 Practical • Thursday • 8:30 - 9:00 Review comparative genomics • 9:00 - 10:00 VectorBase lecture Bioinformatics Tools for Comparative Genomics of Vectors
Bioinformatic Tools for Comparative Genomics of Vectors • Tuesday • Genome sequencing • Strategies • New technologies • ‘Finished’ versus ‘Accessible’ genomes • Genome annotation • Aims and realistic goals • Genefinding • Adding value to the gene predictions (descriptions, xref to other data) • Practical • Artemis practical • IGGI assignments • Wednesday • Thursday Bioinformatics Tools for Comparative Genomics of Vectors
Bioinformatic Tools for Comparative Genomics of Vectors • Tuesday • Wednesday • Comparative genomics • Gene synteny (ortholog/paralog determination) • Feedback to genome annotation • Genetrees • Practical • ACT practical • IGGI assignments • Thursday Bioinformatics Tools for Comparative Genomics of Vectors
Bioinformatic Tools for Comparative Genomics of Vectors • Tuesday • Wednesday • Thursday • VectorBase Bioinformatics Tools for Comparative Genomics of Vectors
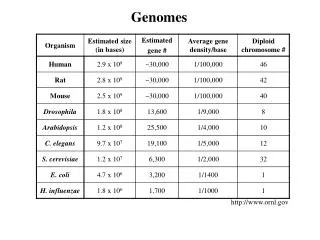
Some terminology • Genome Hereditary information of an organism encoded in the DNA • Chromosome Single large macromolecule of DNA • Contig Single contiguous section of DNA (a set of overlapping DNA segments derived from a single genetic source) • Supercontig (or scaffold) Ordered (and orientated) assembly of contigs • Clone Defined segment of DNA to be used for some purpose • Expressed sequence tag (EST) Short sequence of a transcribed spliced nucleotide sequence. Widely used to identify gene transcripts Bioinformatics Tools for Comparative Genomics of Vectors
Genome size & complexity Increasing complexity Viruses Bacteria Protozoa Inverterbrates Mammals Plants Issues for consideration when sequencing: • DNA source (haplotype issues) • Genome size • Repeat content • Duplications and inversions Issues for consideration when annotating: • Genome size • Repeat content • Splicing (cis and trans) • Genefinding resources (e.g. ESTs) • Likely comparator species Bioinformatics Tools for Comparative Genomics of Vectors
Genome sequencing Sequencing involves: • DNA fragmenting into small pieces • Sequence determination • Assembly into large contiguous sequences Problems occur: • Cloning steps • Bacterial transformation and amplification • Sequencing chemistry (GC compressions, homopolymer runs) • Assembly of repetitive regions Bioinformatics Tools for Comparative Genomics of Vectors
1 2 3 4 5 6 7 8 9 10 11 12 13 Sequencing a Genome Bioinformatics Tools for Comparative Genomics of Vectors
Sequence coverage Most genome sequences are not complete (not finished). Whole Genome Shotguns are referred to as having an X-fold coverage. Low coverage (2x) is sufficient for gene discovery and some regulatory element identification. High coverage (6x) is good for gene annotation. There will still be some missing genes. Finished sequence has no gaps and is presumed to contain all genes. Bioinformatics Tools for Comparative Genomics of Vectors
Sequence strategies Sequencing technologies and strategies for genomic sequencing are constantly changing (improving). • Genomic clones in an ordered ‘clone by clone’ approach • Whole Genome Shotgun (WGS) • Traditional Sanger sequencing long reads • New short-read technologies • Hybrid WGS strategies • Reduced representation WGS using short-read technologies • Mixture of Solexa/454 reads and large-insert clone ends » How big a piece of DNA can we assembly with confidence? Bioinformatics Tools for Comparative Genomics of Vectors
Overlapping BACs 354,510 Tiling set 29,298 4-5x shotgun sequence & computer assembly Draft sequence ……..TAGCTGTGTACGATGATC………. ~15 contigs per clone 4-5x more shotgun Gap closure Problem solving i.e. “Finishing” 1 contig less than one error in 10,000 Finished sequence Sequencing the Human Genome Chromosome 24 Bioinformatics Tools for Comparative Genomics of Vectors
Sequencing data Bioinformatics Tools for Comparative Genomics of Vectors
Output from an automated DNA sequencing machine used by the Human Genome Project to determine the complete human DNA sequence. Bioinformatics Tools for Comparative Genomics of Vectors
1992-1999 Sequencer: gel ABI 373/377 2 or 3 runs per day, 36 to 96 samples 100kb of information per machine per day 80 people 2000 Sequencer: capillary ABI 3700 8 runs per day, 96 samples 400kb of information per machine per day 40 people 2004 Sequencer: capillary ABI 3730xl 15/40 runs per day, 96 samples 2 Mb of information per machine per day 10 people Advanced Technologies Bioinformatics Tools for Comparative Genomics of Vectors
Sequencing by synthesis • Solexa/Illumina sequencing platform. • DNA fragments ligated with adaptors and attached to a flow cell. • Solid state amplification of the sequence (approx. 1000 fold) to form dense (less than 1 micron) spots. • Can achieve very high spot densities (up to 10 million clusters per cm2). • Use labeled reversible terminators and laser excitation to determine incorporated bases • No cloning step improves representation of the genome • No issues relating to homopolymer runs • Read lengths are short, approx. 30-40 bp • Throughput is in the order of 100 Mb per run • 8 samples per flow cell Bioinformatics Tools for Comparative Genomics of Vectors
Solexa sequencing Bioinformatics Tools for Comparative Genomics of Vectors
Pyrosequencing (454) • Nebulized or adapter-ligated DNA fragments are attached to beads • PCR amplification step • Each DNA-bound bead is placed into picotiterplate where the DNA synthesis will take place • Measure incorporation of a nucleotide using the light produced via the luciferase enzyme (nucleotide incorporation releases pyrophosphate which is converted to ATP by ATP sulfurylase and consumed by luciferase producing light). • However, the signal strength for homopolymer stretches is linear only up to eight consecutive nucleotides after which the signal falls-off rapidly • Can deal with high GC composition • No cloning step improves representation of the genomic sequence • Read lengths are approx. 100 bp • Throughput in currently in the order of 20 million bp per run Bioinformatics Tools for Comparative Genomics of Vectors
Comparison of sequencing technologies † New FLX upgrades should increase read lengths to 300bp and throughput to approximately 100 MB Bioinformatics Tools for Comparative Genomics of Vectors
New technologies need new assembly algorithms • Just as the the transition from ‘clone by clone’ approach to Whole Genome Shotgun spawned new algorithms for sequence assembly the increasing use of short-read technologies requires new assembly algorithm developments • Genomics clones (30-300 kb) • Phrap • Chromosomes/Genomes using Sanger long-read technologies (<1000 Mb) • TIGR assembler • ARACHNE • JAZZ • PCAP • Phusion • Genomes using short-read technologies (< 10 Mb) • Velvet • SHARCGS • AbySS Bioinformatics Tools for Comparative Genomics of Vectors
Some terminology • N50 Measure of genome assembly quality. The N50 value is defined as a value for which 50% of the sequenced nucleotides are represented in groups with length greater than this value. Commonly two N50 values are quoted: N50 contig length - a measure of how well individual reads assemble N50 supercontig length - a measure of the general quality of the assembly • Contig Single contiguous section of DNA (a set of overlapping DNA segments derived from a single genetic source) • Supercontig (or scaffold) Ordered (and orientated) assembly of contigs Bioinformatics Tools for Comparative Genomics of Vectors
High-throughput technology leads to lower quality assembled genomes • Few genomes are completely sequenced. The completion and quality assurance needed for bacterial genomes is expensive, for larger eukaryotes even more so. • ‘Finishing’ is the process by which a WGS shotgun assembly is completed (determine the sequence from any physical or sequence gaps) and further polished to remove ambiguities in the base calls and attempt to accurately reflect repetitive regions. • New sequencing technologies provide better representation of the genome (by removing cloning steps) and deeper coverage but are harder to assemble because of the short-read lengths. • People now talk about the ‘accessible’ genome for a species. This simply means the output from a reasonably deep sequence shotgun after assembly and limited (mainly computational) processing and improvements. » Trade off between throughput and product quality. Bioinformatics Tools for Comparative Genomics of Vectors
Sequence substrates • What is the product of a genome assembly? • What is starting material for a genome annotation? • Completed chromosome/genome • Genomic clones • Ordered supercontigs • Unordered supercontigs • Clustered EST sequences† Bioinformatics Tools for Comparative Genomics of Vectors
Sequencing substrates Chromosome Genomic clones Supercontigs Contigs Clustered ESTs Unordered supercontigs Bioinformatics Tools for Comparative Genomics of Vectors
Genome sequencing Annotation quality depends on: • Fragmentation of assembly • Sequencing errors • Poorly represented sequence regions • Extensive simple repeat sequences • Large number of transposon sequences • Haplotype problems • Contaminants (e.g. bacterial or viral sequences) Bioinformatics Tools for Comparative Genomics of Vectors
Genome annotation - the goal! • Defining important features of the genome sequence • Labelling/describing features of the genome • 'Adding value' to the genome sequence • Annotation is an ongoing process • Annotation is almost always incomplete • Set of ‘Best guess’ gene predictions • Short description of the putative function for each prediction • Species/Group dependant catalog of other data types Bioinformatics Tools for Comparative Genomics of Vectors
Annotation from a genome project prospective • Initial ‘first pass’ annotation run prior to publication • Subsequent curation is a collaboration with the community • Focused on protein-coding genes • ‘Best guess’ predictions • Little emphasis on transposons or pseudogenes • Predicting gene loci is more important than getting 100% correct gene structure predictions Bioinformatics Tools for Comparative Genomics of Vectors
Genes Genes Genes Genes Manual v Automated annotation Bioinformatics Tools for Comparative Genomics of Vectors
Manual v Automated: Pros & Cons Speed Coverage * Accuracy * Met’s & STOPs Reproducibility * Bioinformatics Tools for Comparative Genomics of Vectors
Manual (re)annotation - Bridges…… “Paint the Bridge” • Classic “First-pass” annotation strategy • Annotate genomic regions by walking through the chromosome/clone/slice • Comprehensive but slow to deal with problem genes “Painting by numbers” • Identify problem genes by scripts to generate lists for manual appraisal • Responsive to community submissions but only as good as the list generation script Bioinformatics Tools for Comparative Genomics of Vectors
Automated (re)annotation: Ensembl Ensembl builds the bridge anew with each gene build • Responsive to new data • Questions of prediction “churn” Bioinformatics Tools for Comparative Genomics of Vectors
Manual v Automated approaches • Involvement of the community to improve gene prediction accuracy and functional calls • Moderated submissions - (WormBase, FlyBase) • Integration time is dependent on database release cycles • Direct submissions - (VectorBase) • Presentation via DAS onto genome browser • Moderated before integration • Integration time is relatively slow • Indirect submissions - (EMBL/GenBank/DDBJ) • Submissions to public nucleotide databases will get reflected in the genome annotation - eventually! • Processed to protein databases and then integrated Bioinformatics Tools for Comparative Genomics of Vectors
Genome annotation - building a pipeline Genome sequence Map repeats Map ESTs Map Peptides Genefinding nc-RNAs Protein-coding genes Functional annotation Release Bioinformatics Tools for Comparative Genomics of Vectors
Genome annotation - predicting genes Blessed predictions Manual annotations Community submissions (Apollo) (Genewise, Exonerate, Apollo) Similarity predictions Species-specific predictions (Genewise) (Genewise) Canonical predictions Protein family HMMs ncRNA predictions (Genewise) (Rfam) Transcript based predictions ab initio gene predictions (Exonerate) (SNAP) Bioinformatics Tools for Comparative Genomics of Vectors
Annotation Bioinformatics Tools for Comparative Genomics of Vectors