Download

1 / 28
290 likes | 440 Views
Binding features that promote catalysis. co-localization and proper orientation of reacting groups involving different molecules or just a single molecule binding energy is used to pay the entropic cost of bringing reacting groups into the right positions and orientations

E N D
Binding features that promote catalysis • co-localization and proper orientation of reacting groups • involving different molecules or just a single molecule • binding energy is used to pay the entropic cost of bringing reacting groups into the right positions and orientations • stabilization of (and thereby population of) particular configurations of molecules suitable for bond formation/breakage • electrostatic contributions; stabilization of the charged states that develop during a reaction Figure 11-13b
An example of the importance of proximity effects slow reaction fast reaction Figure 11-13b
Understanding catalysis as a lowering of the transition state free energy • articulated by Linus Pauling • recall that providing something that binds a molecule tightly effectively lowers its free energy If the enzymes binding site is most complementary to the transition state (and so lowers the energy of the transition state more than the substrate/products), then the activation free energy is lowered, leading to rate enhancement. Figure 11-13b
The importance of transition state theory • helps explain the mechanisms of catalysis • provides a strategy for designing inhibitors against various enzymes • imagine what the (unstable) t.s. for the reaction must look like • design a (stable) organic mimic that looks similar; this should bind very more tightly to the enzyme than the substrate, and act as a potent inhibitor • an important strategy in drug design Page 339
Example of an inhibitor based on a transition state analogue reaction catalyzed by the enzyme proline racemace transition state analogues sharing some features with the presumed transition state Page 339
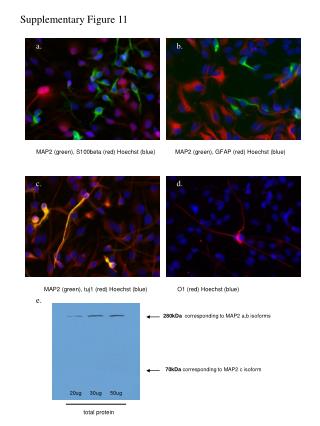
Mechanism of a well-studied enzyme: lysozyme • hydrolyzes b(14) polysaccharides linkages in the bacterial cell wall • resembles an acid catalyzed mechanism in solution • Glu 35 acts as an acid, donating proton to leaving group • carbocation stabilized by • oxonium resonance, promoted by binding the sugar ring in a half-chair configuration • then by covalent bond to Asp 52 • 2nd half of the reaction is the reverse of the first • H20 displaces the leaving sugar • Glu 35 now acts as a base to accept proton from H20, which becomes the attacking group when the Asp 52 leaves • note that ‘double displacement’ leads to retention of the b configuration at the anomeric carbon Page 339
substrate enzyme. note the active site has sub-sites for binding multiple parts of the sustrate Figure 11-16
A planar arrangement of atoms attached to the ring oxygen is required for stabilization of the carbocation that develops at the anomeric carbon when the leaving group leaves. Such a planar configuration is provided by the half-chair ring conformation, which is how the active site binds the substrate. Figure 11-19
Lysozyme mechanism Figure 11-21
Lysozyme mechanism Figure 11-21 part 1
Lysozyme mechanism Figure 11-21 part 2
Lysozyme mechanism Figure 11-21 part 3
Lysozyme mechanism Figure 11-21 part 4
Lysozyme mechanism Figure 11-21 part 5
Lysozyme transition state and t.s. analog Figure 11-22
Mechanism of a well-studied family of enzymes: serine proteases • proteases hydrolyze other proteins • widespread • use a serine nucleophile to attack substrate carbonyl • because the serine nucleophiles in these enzymes are so nucleophilic, they can be ‘killed’ (i.e. irreversibly modified by a covalent bond) by ‘suicide inhibitors’ Page 339
Mechanism of a well-studied family of enzymes: serine proteases • specificities of different serine proteases vary (e.g. trypsin, chymotrypsin, etc.) • specificity pockets in the binding site for recognizing certain amino acids in the substrate proteins Page 339
Mechanism of a well-studied family of enzymes: serine proteases • Mechanism: • serine is promoted as a nucleophile by deprotonation by nearby histidine side chain (whose resulting positive charge is stabilized by a nearby aspartate). This is the famous ‘catalytic triad’ • departure of C-terminal fragment of substrate • a covalent intermediate is formed in which the N-terminal fragment of the substrate is attached to the enzyme as an acyl-enzyme intermediate • 2nd step is a direct repeat, but with H20 replacing the serine as the nucleophile Page 339
Serine proteases: the catalytic triad note that the participating residues are coming from different parts of the enzyme sequence Figure 11-26
Similar catalytic triads have been discovered in apparently unrelated enzymes, having different overall three-dimensional structures and different orderings of the triad residues, suggesting that this particularly useful arrangement of catalytic residues has evolved independently multiple times (i.e. ‘convergent evolution’) Figure 11-28
Serine protease mechanism Figure 11-29
Serine protease mechanism Figure 11-29 part 1
Serine protease mechanism Figure 11-29 part 2
Serine protease mechanism Figure 11-29 part 3
Serine protease mechanism Figure 11-29 part 4
Serine protease mechanism Figure 11-29 part 5
Serine proteases: features for transition state stabilization • ‘oxyanion hole’ to stabilize negative charge on tetrahedral intermediate • H-bonds to stabilize distorted protein backbone Figure 11-30a
One biologically important use of proteases: to cleave inactive proenzymes (or zymogens) to produce the mature, active form of an enzyme In some cases, such cleavages act one after another on a series of enzymes. This leads to a ‘cascade’, which can produce a geometric explosion of enzymatic activity (which may be needed in response to catastrophic events). The well-known blood clotting cascade is shown. Box 11-4c