Download

1 / 30
300 likes | 525 Views
Identifying Structural Motifs in Proteins. Rohit Singh Joint work with Mitul Saha. The Big Picture: small motifs. Active Sites are preserved across proteins with similar functions. The Big Picture: large motifs. Even bigger motifs are often conserved. . Oh, BTW….

E N D
Identifying Structural Motifs in Proteins Rohit Singh Joint work with Mitul Saha
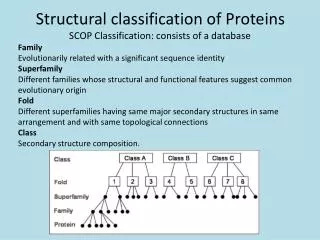
The Big Picture: small motifs Active Sites are preserved across proteins with similar functions
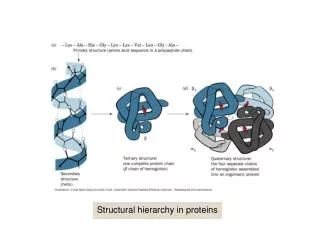
The Big Picture: large motifs Even bigger motifs are often conserved.
Oh, BTW… There are two different issues here: • Find the best match for the motif in the protein • Extensively studied in vision/graphics • Is the match “significant” ? • For small motifs a good match is more likely • What is probability of a match against a random protein being this good ? (cf. BLAST)
What’s in it for a CS guy ? • The problem of matching two point-sets has many applications • Most current algorithms geared towards points that are indistinguishable (e.g. points on a mesh) • There are few rigorous results on the significance of matches
So what have we done ? • Towards a more rigorous approach for scoring the quality of a match (between motif and protein) • Provide a method that is capable of finding the optimum match based on these criteria
Problem Description • Given a motif and a protein, for each point in the motif, find a corresponding point in the protein. • Given these correspondences, find the best transformation (rotation and translation only) of the motif that aligns it to the protein. • Optimize over all possible correspondences
Oh, BTW… • Given two sets of k points, easy to find the optimal rotation and translation that minimizes the least sum-of-squared error (also RMSD). • Boils down to finding the largest eigenvalue of a 4x4 matrix.
Previous Work • Brute Force approach: match edges of same length. • Geometric Hashing: Pennec & Ayache, Bioinformatics, 1998
What is missing ? • Ad hoc: Try to minimize a quantity that is only indirectly related to the least square error or RMSD. • Hard to evaluate the quality of partial matches • Brute Force methods infeasible for larger motifs • Geometric Hashing requires significant preprocessing
Estimating the error Model the alignment problem as a regression problem: Y = model set (protein) T = data set (motif) g = transformation (rot+trans) • Which error criterion to use ? • Least Mean Squared Error (also RMSD) • LSE is not good when you have outliers. • what to do ?
Robust error estimation • LSE: larger error terms have disproportionate influence. • Use a function to reduce the effect of larger error terms (M-estimators)
Its an optimization problem! Consider the case of full matching: Domain: set of all possible correspondences between points on the motif and points on the protein Range: given a particular set of corresponding points, the minimum error in aligning those point sets. Goal: find the global minimum of this function!
Looking for global minimum Our approach: • Prune the search space to a small and plausible sub-space • Find (most) of the local minima in this sub-space quickly • Choose the minimum over these local minima
Finding local minima is easy:ICP Iterative Closest Point (Besl-McKay):
ICP contd… • ICP is guaranteed to converge to a local minimum • But depends a lot on initial seeding • Convergence is quick: ~4-5 iterations ICP movie
Pruning the search space • Every point in motif/protein has some features: • Amino acid type, element type, sec. structure, hydrophobic/polar, ‘substitutable’ • Assume: a point with feature X can only match another point with feature X (or {Y,Z,W}) • Assume: some features are more frequent than others
Our Approach • Find the feature that is least frequent in protein. • For each occurrence of the feature: • Seed ICP appropriately. Find local minimum. • Look around a few more times • Return the best answer you have
Observations • Will always find a perfect match, if it exists. • Moreover, will find such a match quickly. • The error is directly interpretable in RMSD terms
…contd Trypsin active site against Trypsin like proteins
…contd Trypsin active site against kinases
What about partial matching ? • Basic idea is the same: pruning+ICP • Replace least squared error estimates by M-estimator based errors. Problem: How to find the optimal rotation/translation that minimizes this new variety of error criterion? Answer: weighted LSE ? Is there a better way ?
RANSAC Choice of the parameters has statistical justification
…contd Data for distorted trypsin active site against ten different trypsins:
Future Work • Test on larger motifs: secondary structure elements • Choice of better features • A theoretical guarantee about the quality of results • Explore different criteria for partial matching