Download

1 / 33
390 likes | 1.2k Views
InnoMol Proteomics Workshop April 8, 2014. Principles of Shotgun Proteomics and Proteogenomics. Boris Ma č ek Proteome Center Tuebingen. General MS-based proteomics workflow. Aebersold R and Mann M. 2003 . Nature 422 : 198-207. Principle of protein database search.

E N D
InnoMol Proteomics Workshop April 8, 2014 Principles of Shotgun Proteomics and Proteogenomics Boris Maček Proteome Center Tuebingen
General MS-based proteomics workflow Aebersold R and Mann M.2003.Nature 422: 198-207
Principle of protein database search A L K G A S Intensity Translated Genomic Sequence Theoretical Spectra for Proteins Intensity Intensity Intensity Intensity m/z m/z m/z m/z m/z Theoretical spectra that fall into the defined mass range. Intensity Each of them is compared to our fragment Ion spectra. m/z Intensity m/z Database
Principle of protein database search >sp|P31946|1433B_HUMAN 14-3-3 protein beta/alpha OS=Homo sapiens GN=YWHAB PE=1 SV=3 MTMDKSELVQKAKLAEQAERYDDMAAAMKAVTEQGHELSNEERNLLSVAYKNVVGARRSSWRVISSIEQKTERNEKKQQMGKEYREKIEAELQDICNDVLELLDKYLIPNATQPESKVFYLKMKGDYFRYLSEVASGDNKQTTVSNSQQAYQEAFEISKKEMQPTHPIRLGLALNFSVFYYEILNSPEKACSLAKTAFDEAIAELDTLNEESYKDSTLIMQLLRDNLTLWTSENQGDEGDAGEGEN >sp|P62258|1433E_HUMAN 14-3-3 protein epsilon OS=Homo sapiens GN=YWHAE PE=1 SV=1 MDDREDLVYQAKLAEQAERYDEMVESMKKVAGMDVELTVEERNLLSVAYKNVIGARRASWRIISSIEQKEENKGGEDKLKMIREYRQMVETELKLICCDILDVLDKHLIPAANTGESKVFYYKMKGDYHRYLAEFATGNDRKEAAENSLVAYKAASDIAMTELPPTHPIRLGLALNFSVFYYEILNSPDRACRLAKAAFDDAIAELDTLSEESYKDSTLIMQLLRDNLTLWTSDMQGDGEEQNKEALQDVEDENQ >sp|P62258-2|1433E_HUMAN Isoform SV of 14-3-3 protein epsilon OS=Homo sapiens GN=YWHAE MVESMKKVAGMDVELTVEERNLLSVAYKNVIGARRASWRIISSIEQKEENKGGEDKLKMIREYRQMVETELKLICCDILDVLDKHLIPAANTGESKVFYYKMKGDYHRYLAEFATGNDRKEAAENSLVAYKAASDIAMTELPPTHPIRLGLALNFSVFYYEILNSPDRACRLAKAAFDDAIAELDTLSEESYKDSTLIMQLLRDNLTLWTSDMQGDGEEQNKEALQDVEDENQ >sp|Q04917|1433F_HUMAN 14-3-3 protein eta OS=Homo sapiens GN=YWHAH PE=1 SV=4 MGDREQLLQRARLAEQAERYDDMASAMKAVTELNEPLSNEDRNLLSVAYKNVVGARRSSWRVISSIEQKTMADGNEKKLEKVKAYREKIEKELETVCNDVLSLLDKFLIKNCNDFQYESKVFYLKMKGDYYRYLAEVASGEKKNSVVEASEAAYKEAFEISKEQMQPTHPIRLGLALNFSVFYYEIQNAPEQACLLAKQAFDDAIAELDTLNEDSYKDSTLIMQLLRDNLTLWTSDQQDEEAGEGN >tr|F2Z3E5|F2Z3E5_HUMAN Hydroxyacid-oxoacidtranshydrogenase, mitochondrial OS=Homo sapiens GN=ADHFE1 PE=4 SV=1 MAAAARARVAYLLRQLQRAACQCPTHSHTYSQDGCFKY >tr|Q5SS58|Q5SS58_HUMAN MHC class I polypeptide-related sequence A OS=Homo sapiens GN=MICA PE=4 SV=2 MGQRDQGLDRERKGPQDDPGSYQGPERRNFLKEDAMKTKTHYHAMHADCLQELRRYLESGVVLRRTVPPMVNVTRSEASEGNITVTCRASSFYPRNIILTWRQDGVSLSHDTQQWGDVLPDGNGTYQTWVATRICRGEEQRFTCYMEHSGNHSTHPVPSGKVLVLQSHWQTFHVSAVAAGCCYFCYYYFLCPLL >tr|Q5T409|Q5T409_HUMAN Disrupted in schizophrenia 1 OS=Homo sapiens GN=DISC1 PE=2 SV=1 MPGGGPQGAPAAAGGGGVSHRAGSRDCLPPAACFRRRRLARRPGYMRSSTGPGIGFLSPAVGTLFRFPGGVSGEESHHSESRARQCGLDSRGLLVRSPVSKSAAAPTVTSVRGTSAHFGIQLRGGTRLPDRLSWPCGPGSAGWQQEFAAMDSSETLDASWEAACSDGARRVRAAGSLPSAELSSNSCSPGCGPEVPPTPPGSHSAFTSSFSFIRLSLGSAGERGEAEGCPPSREAESHCQSPQEMGAKAASLDGPHEDPRCLSRPFSLLATRVSADLAQAARNSSRPERDMHSLPDMDPGSSSSLDPSLAGCGGDGSSGSGDAHSWDTLLRKWEPVLRDCLLRNRRQMEVISLRLKLQKLQEDAVENDDYDKAETLQQRLEDLEQEKISLHFQLPSRQPALSSFLGHLAAQVQAALRRGATQQASGDDTHTPLRMEPRLLEPTAQDSLHVSITRRDWLLQEKQQLQKEIEALQARMFVLEAKDQQLRREIEEQEQQLQWQGCDLTPLVGQLSLGQLQEVSKALQDTLASAGQIPFHAEPPETIRSLQERIKSLNLSLKEITTKVCMSEKFCSTLRKKVNDIETQLPALLEAKMHAISGNHFWTAKDLTEEIRSLTSEREGLEGLLSKLLVLSSRNVKKLGSVKEDYNRLRREVEHQETAYETSVKENTMKYMETLKNKLCSCKCPLLGKVWEADLEACRLLIQSLQLQEARGSLSVEDERQMDDLEGAAPPIPPRLHSEDKRKTPLKESYILSAELGEKCEDIGKKLLYLEDQLHTAIHSHDEDLIHSLRRELQMVKETLQAMILQLQPAKEAGEREAAASCMTAGVHEAQA A L K G A S MaxQuant Software Translated Genomic Sequence Theoretical Spectra for Proteins Intensity m/z Homo Sapiens Reference Proteome 71,434 entries (20,246 reviewed proteins) (51,188 un-reviewed) Database
MS instrumentation in proteomics Aebersold R and Mann M.2003.Nature 422: 198-207
Column (75 µm)/spray tip (8 μm) Reverse-phase C18 beads, 3 μm Coupling LC to MS for complex mixture analysis Nanoflow LC/MS interface set-up: Proxeon Easy nLC nanoflow LC System LTQ-Orbitrap No precolumn or split! 12-15 cm Sample Loading:~700 nl/min Gradient elution:~200 nl/min Platin-wire 2.0 kV
Coupling LC to MS for complex mixture analysis BSA tryptic in-solution digest 50 fmol on column
LTQ-Orbitrap (2005) Octopole coll. cell Linear ion trap (LTQ) Source C-Trap Orbitrap LTQ-FT MS/MS optimized scan cycle: → peptide mass measurement → peptide sequencing Orbitrap-MS MS-Full Scan MS2 MS2 MS2 MS2 MS2 LTQ-MS 0 300 600 900 1200 1500 1800 Time [msec]
Acquisition speed LTQ Orbitrap XL LTQ Orbitrap Velos □ CID Identified + CID Not Iidentified
Acquisition speed # of MS/MS Scans
Stable Isotope Labeling byAmino Acids in Cell Culture (SILAC) ”normal AA” ”heavy AA” Lys-12C6 Lys-13C6 Resting cells Treated (drug, GF) Combine and lyse, protein purification or fractionation Proteolysis (trypsin, Lys-C, etc.) Quantitation and identification by MS (nanoscale LC-MS/MS)
Current research at the PCT • Proteogenomics • B. subtilis, E. coli (Krug et al, 2011, Mol Bosystems; 2013 MCP) • Pristionchus pacificus (Borchert et al, 2010, Genome Res) • cancer cell lines/tissues • Proteomics for systems biology • In-depth sequencing and quantitation of model organisms (B.subtilis, • E.coli, S. pombe, A. thaliana) (Soufi et al, 2010, J Prot Res; Schütz et al, 2011, Plant Cell; Soufi et al, 2012, Curr Opinion Microbiol; Soares et al, 2013, JPR) • Phosphoproteomics • targets of Aurora kinase in S. pombe (Koch et al, 2011, Science Signaling) • targets of protein kinase D in human cells (Franz-Wachtel et al., 2012, MCP) • targets of S/T/Y kinases and phosphatases in B.subtilis and E.coli • Protein modifications • ubiquitylation (Ikeda et al, 2011, Nature) • lysine acetylation (Carpy et al., in preparation) • Clinical proteomics • genetic rescue of Fragile X phenotype in FMR1 KO mice
E. coli: Replicate 1 and 2 *in all phases of growth Soufi et al. in preparation
Biological reproducibility Soufi et al. in preparation
Proteome dynamics during growth Soufi et al. in preparation
Dynamics of stress proteins during growth Soufi et al. in preparation
Estimation of absolute copy numbers T6 T5 T7 T4 OD 600 T3 UPS standard (iBAQ) T2 T1 1800 5760 Time (min) Soufi et al. in preparation
Summary of absolutely quantified proteins Soufi et al. in preparation
Most abundant Proteins (ES) Soufi et al. in preparation
Dynamic range of protein abundance Count Blue: All proteins Red: Membrane proteins Log2 Protein Copy Number Soufi et al. in preparation
Proteogenomics • Application of tandem mass spectrometry to genome re-annotation • Search MS/MS spectra against a database containing the complete genome • translated in 6 reading frames
Problem: database size and structure „Ususal“ Proteomics applications • Incompatibility with some data processing programs • Long search times • Decreased sensitivity of database search • Unequal target and decoy search spaces • Most translated frames are in fact decoy sequences • Overestimation of the FDR Predicted ORFs REV_Predicted ORFs Proteogenomics applications Predicted ORFs Frame1 Frame2 Frame3 Frame4 Frame5 Frame6 REV_Predicted ORFs REV_Frame1 REV_Frame2 REV_Frame3 REV_Frame4 REV_Frame5 REV_Frame6
Proteogenomics of E. coli • Model Gram-negative bacterium • Small (4.6 Mb) and well characterized genome • ~4,300 protein coding genes (manually annotated and reviewed) • Comprehensive high accuracy MS dataset comprising >42,000 unique peptide sequences from >2,600 proteins • Hypothesis: genome annotation approaches completeness • Assessment of general properties of a simple proteogenomic experiment
Proteogenomics of E. coli 1.9M peptide mass spectra
Proteogenomics of E. coli PEP = 4.02E-08 PP = 0.9999 Annotated genes Detected peptides Six-frame ORFs A B ybdz fes fepa fes Annotated genes Detected peptides Six-frame ORFs MFEVTFWWRDPQGSEEY... VGSESWWQSK TWGYGVTALKVGSESWWQSKHGPEWQRLNDEMFEVTFWWRDPQGSEEY... PEP = 0.027976 PP = 0.9504 C D yhjb yhja tref Position (Mb) Position (Mb) tref MLNQKIQNPNPDELMIEVDLCYELDPYELKLDEMIEAEP... KPPQIRISL ...NAVFKPPQIRISL LATNFGGWILMLNQKIQNPNPDELMIEVDLCYELDPYELKLDEMIEAEP... Krug et al. Mol Cell Proteomics, 2013
Majority of Novel Peptides are False Positives Krug et al. Mol Cell Proteomics, 2013
Assessment of Processing Workflows Krug et al. Mol Cell Proteomics, 2013
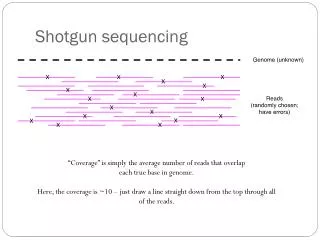
Deep Proteome Coverage of Escherichia coli MS/MS scans Mean: 20 scans Median: 7 scans 0 50 100 150 20-fold base coverage of 27.5% genome sequence Krug et al. Mol Cell Proteomics, 2013
Conclusions • proteomics reaches analytical capacity to identify and quantify all gene • products in microorganisms grown in culture • several regulatory protein modifications (e.g. S/T/Y-phosphorylation, lysine • acetylation) can routinly be analyzed on a global scale • many challenges ahead: • analysis of H/D-phosphorylation • analysis of environmental samples • coverage of genome/protein sequence by detected peptides • future developments: • faster MS/MS acquisition • smarter acquisition software • large-scale targeted proteomics • metaproteomics and individual proteomics
Acknowledgements Proteome Center Tuebingen Boumediene Soufi Nelson C. Soares Philipp Spät Karsten Krug Alejantro Carpy Sasa Popic Silke Wahl Funding