Download
1 / 3
30 likes | 121 Views
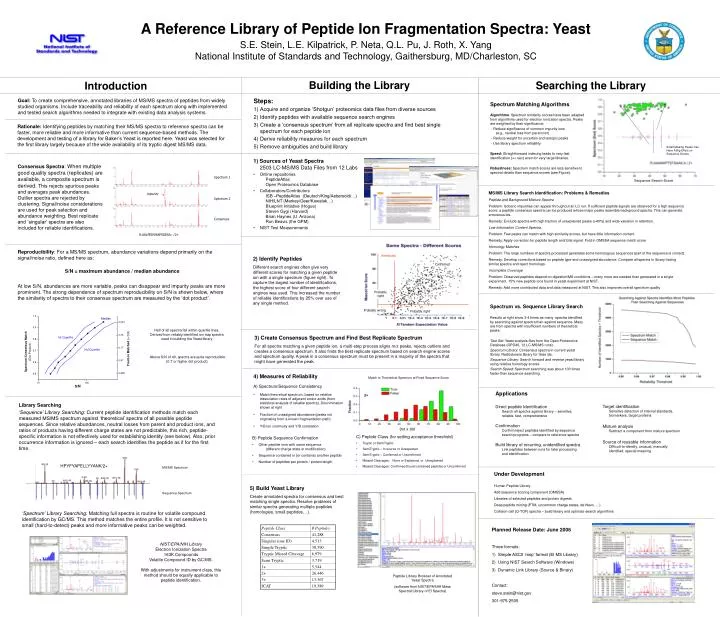
thresholds. Confirmed. Spectrum 1. impurity. Spectrum 2. Probably right. Consensus. Probably wrong. Probably right. R.AALPEDVNAPSGEAA.- /2+. HPYFYAPELLYYANK/2+. MS/MS Spectrum. Sequence Spectrum. Building the Library. Searching the Library. Introduction. Steps:

E N D
thresholds Confirmed Spectrum 1 impurity Spectrum 2 Probably right Consensus Probably wrong Probably right R.AALPEDVNAPSGEAA.- /2+ HPYFYAPELLYYANK/2+ MS/MS Spectrum Sequence Spectrum Building the Library Searching the Library Introduction • Steps: • Acquire and organize ‘Shotgun’ proteomics data files from diverse sources • Identify peptides with available sequence search engines • Create a ‘consensus spectrum’ from all replicate spectra and find best single spectrum for each peptide ion • Derive reliability measures for each spectrum • Remove ambiguities and build library Goal: To create comprehensive, annotated libraries of MS/MS spectra of peptides from widely studied organisms. Include traceability and reliability of each spectrum along with implemented and tested search algorithms needed to integrate with existing data analysis systems. Spectrum Matching Algorithms Algorithms: Spectrum similarity scores have been adapted from algorithms used for electron ionization spectra. Peaks are weighted by their significance: - Reduce significance of common impurity ions (e.g., neutral loss from parent ion) - Reduce weight for uncertain and isotopic peaks - Use library spectrum reliability Speed: Straightforward indexing leads to very fast identification (<< sec) even for very large libraries. Robustness: Spectrum match scores are less sensitive to spectral details than sequence scores (see Figure). A Reference Library of Peptide Ion Fragmentation Spectra: YeastS.E. Stein, L.E. Kilpatrick, P. Neta, Q.L. Pu, J. Roth, X. YangNational Institute of Standards and Technology, Gaithersburg, MD/Charleston, SC Small Missing Peaks Can Have A Big Effect on Sequence Scores • 1) Sources of Yeast Spectra2503 LC-MS/MS Data Files from 12 Labs • Online repositories PeptideAtlas Open Proteomics Database • Collaborators/Contributors ISB –PeptideAtlas (Deutsch/King/Aebersold/…) NIH/LNT (Markey/Geer/Kowalak…) Blueprint Initiative (Hogue) Steven Gygi (Harvard) Brian Haynes (U. Arizona) Ron Beavis (the GPM) • NIST Test Measurements Consensus Spectra: When multiple good quality spectra (replicates) are available, a composite spectrum is derived. This rejects spurious peaks and averages peak abundances. Outlier spectra are rejected by clustering. Signal/noise considerations are used for peak selection and abundance weighting. Best replicate and ‘singular’ spectra are also included for reliable identifications. Sequence Search Score MS/MS Library Search Identification: Problems & Remedies Peptide and Background Mixture Spectra Problem: Isobaric impurities can appear throughout an LC run. If sufficient peptide signals are observed for a high sequence score, a peptide consensus spectra can be produced whose major peaks resemble background spectra. This can generate erroneous ids. Remedy: Exclude spectra with high fraction of unexplained peaks (>40%) and wide variation in retention. Low Information Content Spectra Problem: Few peaks can match with high similarity scores, but have little information content Remedy: Apply correction for peptide length and total signal. Fold in OMSSA sequence match score Homology Matches Problem: The large numbers of spectra processed generates some homologous sequences (part of the sequence is correct). Remedy: Develop corrections based on peptide type and unassigned abundance. Compare all spectra in library having similar spectra and reject homologs. Incomplete Coverage Problem: Observed peptides depend on digestion/MS conditions – many more are needed than generated in a single experiment. 15% new peptide ions found in yeast experiment at NIST. Remedy: Add more contributed data and data measured at NIST. This also improves overall spectrum quality Reproducibility: For a MS/MS spectrum, abundance variations depend primarily on the signal/noise ratio, defined here as: S/N = maximum abundance / median abundance At low S/N, abundances are more variable, peaks can disappear and impurity peaks are more prominent. The strong dependence of spectrum reproducibility on S/N is shown below, where the similarity of spectra to their consensus spectrum are measured by the ‘dot product’. 2) Identify Peptides Different search engines often give very different scores for matching a given peptide ion with a single spectrum (figure right). To capture the largest number of identifications, the highest score of four different search engines was used. This increased the number of reliable identifications by 25% over use of any single method. Spectrum vs. Sequence Library Search Results at right show 3-4 times as many spectra identified by searching against spectra than against sequence. Many are from spectra with insufficient numbers of theoretical peaks. Test Set: Yeast analysis files from the Open Proteomics Database (OPD40, 12 LC-MS/MS runs). Spectrum Library: Consensus spectra in current yeast library. Radiodurans library for false ids. Sequence Library: Search forward and reverse yeast library using relative homology scores. Search Speed: Spectrum searching was about 100 times faster than sequence searching. Rationale: Identifying peptides by matching their MS/MS spectra to reference spectra can be faster, more reliable and more informative than current sequence-based methods. The development and testing of a library for Baker’s Yeast is reported here. Yeast was selected for the first library largely because of the wide availability of its tryptic digest MS/MS data. Half of all spectra fall within quartile lines. Derived from reliably identified ion trap spectra used in building the Yeast library 3) Create Consensus Spectrum and Find Best Replicate Spectrum For all spectra matching a given peptide ion, a multi-step process aligns m/z peaks, rejects outliers and creates a consensus spectrum. It also finds the best replicate spectrum based on search engine scores and spectrum quality. A peak in a consensus spectrum must be present in a majority of the spectra that might have generated the peak. Above S/N of 40, spectra are quite reproducible (0.7 or higher dot product). • 4) Measures of Reliability • A) Spectrum/Sequence Consistency • Match theoretical spectrum, based on relative dissociation rates of adjacent amino acids (from statistical analysis of reliable spectra). Discrimination shown at right • Fraction of unassigned abundance (peaks not originating from a known fragmentation path) • Y/B ion continuity and Y/B correlation Match to Theoretical Spectrum at Fixed Sequence Score Applications Direct peptide IdentificationSearch all spectra against library – sensitive, reliable, fast, comprehensive ConfirmationConfirm/reject peptides identified by sequence search programs – compare to reference spectra Build library of recurring, unidentified spectraLink peptides between runs for later processing and identification Target identificationSensitive detection of internal standards, biomarkers, target proteins Mixture analysisSubtract a component from mixture spectrum Source of reusable informationDifficult-to-identify, unusual, manually identified, special meaning Library Searching ‘Sequence’ Library Searching: Current peptide identification methods match each measured MS/MS spectrum against ‘theoretical’ spectra of all possible peptide sequences. Since relative abundances, neutral losses from parent and product ions, and ratios of products having different charge states are not predictable, this rich, peptide-specific information is not effectively used for establishing identity (see below). Also, prior occurrence information is ignored – each search identifies the peptide as if for the first time. Dot x 100 • C) Peptide Class (for setting acceptance threshold) • Tryptic or SemiTryptic • SemiTryptic – In source or Unexpected • SemiTryptic – Confirmed or Unconfirmed • Missed Cleavages: None or Explained, or Unexplained • Missed Cleavages: Confirmed (found contained peptide) or Unconfirmed • B) Peptide Sequence Confirmation • Other peptide ions with same sequence (different charge state or modification) • Sequence contained in (or contains) another peptide • Number of peptides per protein / protein length Under Development Human Peptide Library Add sequence scoring component (OMSSA) Libraries of selected peptides and protein digests Deep peptide mining (PTM, uncommon charge states, de Novo, …) Collision cell (Q-TOF) spectra – build library and optimize search algorithms 5) Build Yeast Library Create annotated spectra for consensus and best matching single spectra. Resolve problems of similar spectra generating multiple peptides (homologies, small peptides, ..). ‘Spectrum’ Library Searching: Matching full spectra is routine for volatile compound identification by GC/MS. This method matches the entire profile. It is not sensitive to small (hard-to-detect) peaks and more informative peaks can be weighted. • Planned Release Date: June 2006 • Three formats: • Simple ASCII ‘msp’ format (EI MS Library) • Using NIST Search Software (Windows) • Dynamic Link Library (Source & Binary) • Contact: • steve.stein@nist.gov • 301-975-2505 NIST/EPA/NIH Library Electron Ionization Spectra 163K Compounds Volatile Compound ID by GC/MS. With adjustments for instrument class, this method should be equally applicable to peptide identification. Peptide Library Browser of Annotated Yeast Spectra (software from NIST/EPA/NIH Mass Spectral Library of EI Spectra)
Goal • Overview • Background • +S/N • +Consensus Spectra • +Spectrum Variability • Completeness • Library Searching • Library Building • Sources – list of contributors • Multiple SE – show scatter diagram • Quality Control • Individual Spectrum features • Relative spectrum features (use rest of lib) • Common sequence (charge, mod, missed and semitryptic cleavage • # peps in protein • Classes of Peptides – table with thresholds • False IDs and Low Quality Spectra • Random match errors – handle with reverse and spectrum modeling • Impurities – consensus spectra, set threshold • Low S/N – dropped peaks in consensus – use best of replicates • One hit wonders – accept singular spectra • Homology matches – selected protein digests to set thresholds • Library Search Algorithms • Show equation with corrections • Performance testing/optimization • Library Search Concerns • Peptide length and spectrum complexity dependences • Propagation of errors – false ids of common impurities • Special Features • Unusual peptides • Biomarkers • Plans • Acknowledgements
Spectrum/Sequence Scores Vary More than Spectrum/Spectrum Scores Small Missing Peaks Can Have A Big Effect on Sequence Scores Sequence score






















