Download

1 / 10
100 likes | 282 Views
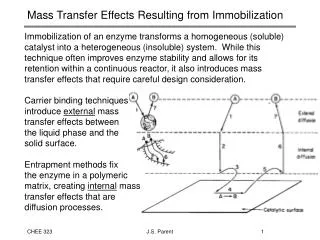
Disorders Resulting from Defects in Lysosomal Function. Characteristics of Lysosomes. Lysosome is a heterogenous organelle (异质性细胞器) : Primary lysosome (初级溶酶体) Second lysosomes (次级溶酶体) heterophagic (异噬溶酶体) autophagic (自噬溶酶体) Residual body (残余小体). Primary Lys. Second Lys.

E N D
Characteristics of Lysosomes Lysosome is a heterogenous organelle(异质性细胞器): • Primary lysosome(初级溶酶体) • Second lysosomes(次级溶酶体) • heterophagic (异噬溶酶体) • autophagic (自噬溶酶体) • Residual body (残余小体) Primary Lys Second Lys
The Functions of Lysosomes • Lysosomes are involved in three major cell functions: ① phagocytosis(吞噬) A summary of the phagocytic pathway
② autophagy(自吞) Electron micrograph of a mitochondrion and peroxisome(过氧化物酶体)enclosed in a double membrane wrapper derived from the ER.This autophagic vacuole would have fused with a lysosome and its contents digested.
Disorders Resulting from Defects in lysosomal Function • I-cell disease : • Many cells in these patients contain lysosomes that are bloated with undergraded materials. • When fibroblasts from these patients were studied in culture ,it was found that lysosomal enzymes are synthesized at normal levels but are secreted into the medium and not targeted to lysosomes.
The I-cell defect was soon traced to the deficiency of an enzyme(N-acetyglucosamine phosphotransferase)required for mannose phosphorylation. The secreted enzymes lacked the mannose phosphate residue that are present on the corresponding enzymes of cells from normal individuals.
Pompe disease: a fatal inherited condition in the absence ofα-glucosidase ,undigested glycogen accumulated in lysosomes,causing swelling of the organelles and irreversible damage to the cells and tissues Diseases of this type, characterized by the deficiency of a single lysosomal enzyme and the corresponding accumulation of undergraded substrate,are called lysosomal storage diseases
Tay-Sachs disease: the best –studied lysosomal storage diseases It results from a deficiency of the enzyme β-N-hexosaminidase A, an enzyme that degrades the ganglioside GM2 GM2 is a major component of the membranes of brain cells ,and in the absence of the hydrolytic enzyme ,the ganglioside accumulates in the bloated lysosomes of brain cells,causing dysfunction In its severe form ,which strikes during infancy ,the disease is chacterized by progressive mental and motor retardation, as well as skeletal ,cardiac,and resiratory abnormalities