Download

1 / 45
770 likes | 1.53k Views
The Lysosome and Lysosomal Storage Disorders (LSD). Part I: Historical, Physiological and Pathological Considerations. Serge Melançon, MD January 2010. Synopsis (Part 1A). Historical notes and scientific partners The endosomal-lysosomal system The ‘synthetic’ pathway

E N D
The Lysosome and Lysosomal Storage Disorders (LSD) Part I: Historical, Physiological and Pathological Considerations Serge Melançon, MD January 2010
Synopsis (Part 1A) • Historical notes and scientific partners • The endosomal-lysosomal system • The ‘synthetic’ pathway • The ‘endocytotic’ pathway • Retrograde transport
Synopsis (Part 1B) • Cofactors for lysosomal enzymes • Secretion-recapture pathway • Plasma membranes and lipid rafts • Molecular genetics • Microglia • Blood-brain barrier (BBB) • Causes of lysosomal storage • Residual enzymatic activity • Effects of lysosomal storage
The discovery of lysosomes Why? In 1955, Belgian scientist Christian de Duve observed that the cells released an enzyme called acid phosphatase in much larger amounts when they were repeatedly frozen and thawed before centrifugation de Duve C (1975) Exploring cells with a centrifuge. Science 189, 186-194
To explain this phenomenon, de Duve suggested that the digestive enzyme must have been encased in some sort of membrane-bound organelle within the cell.
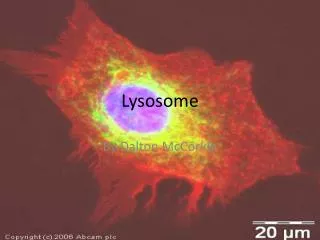
After estimating the probable size of the lysosomes, he was able to identify the organelle in images produced with an electron microscope Lysosomes are spherical organelles contained by a single layer membrane, though their size and shape vary to some extent.
Using brightfield or phase contrast microscopy, the size of these cellular organelles range from 0.25 to 0.5 mm Several hundred lysosomal structures can be seen in the above phase micrograph
"Lysosome" was the name given because of these enzymes' ability to "lyse" the cell • Initially referred to as "suicide bags" since one of the functions of lysosomes is to rupture when the cell dies, • Rupturing releases hydrolytic enzymes that digest all parts of the cell, including proteins, DNA, RNA, carbohydrates, lipids and cellulose.
CELL GEOGRAPHY 101
Rough ER The RER is where hydrolytic enzymes are manufactured before being transported to the Golgi apparatus (complex), where they undergo additional processing and are transformed from an inactive to an active state fig.cox.miami.edu/ ~cmallery/150/cells/ER.jpg
Golgi complex • The Golgi is the distribution • and shipping department for • the cell's chemical products • It plays three important roles 1 Modification of complex molecules (such as proteins) by the addition of sugars (glycosylation) 2 Proteolysis of peptide molecules which makes them become active 3 Sorting of molecules for either, transport out of the cell, incorporation in the cell membrane, or transport to another part of the cell
The Golgi is divided into three functionally separate areas. • The trans-Golginetwork (TGN) (closest to the cell membrane) which performs proteolysis and sorts molecules for their destination • The medial Golgi which adds sugars to both lipids (fats) and peptides (proteins) • The cis face (closest to the nucleus and the ER) receives transport vesicles from the smooth ER
Mitochondria The mitochondria are essential in the production of energy (via adenosine triphosphase; ATP) and lipid biosynthesis io.uwinnipeg.ca/~simmons/ 1115/cm1503/Image110.gi
Peroxisomes contain enzymes That convert hydrogen peroxide to water and render potentially toxic substances safe for the cell And also initiate the production of phospholipids, which are typically used to build up cell membranes.
The endosomal–lysosomal system • The lysosome is one component of a series of unconnected (?) intracellular organelles, collectively known as the endosomal–lysosomal system or vacuolar apparatus (De Duve and Wattiaux,1966) • The various components of the system were described over 30 years ago by Novikoff (1973)
The endosomal–lysosomal system • The main components are • the early endosome, situated at the cell periphery, • the late endosome, which is perinuclear, and • the lysosome. • They form a chain that is responsible for • the trafficking and digestion of endocytosed molecules • and participate actively in sorting and recycling
The lysosome (De Duve 1955) ‘terminal compartment of the system’ • Characteristics • a membrane, • a low internal pH, • vesicles containing hydrolytic enzymes. • The membrane contains • transport systems to carry particles between lumen and cites, • an electrogenic proton pump (V-type H+-ATPase, Arai et al, 1993). • several membrane proteins of uncertain function (Eskelinen et al, 2003).
The lysosome micro.magnet.fsu.edu /.../ lysosomesfigure1.jpg
Role of the lysosome • Substrate breakdown using endocytosis • Secretion of its content after fusion with the plasma membrane (Luzio et al, 2000). • Phagocytosis of bacteria and cellular debris to form phagolysosomes. • Calcium-regulated exocytosis for membrane repair (Reddy et al, 2001)
Synthesis and trafficking of lysosomal enzymes (the 'synthetic' pathway) • Glycoproteins synthesized in the rough endoplasmic reticulum (RER) (At this early stage they are inactive) • Translocate through the ER membrane with the help of N-terminal signal sequences • Once in the lumen of the ER, undergo N-glycosylation and lose the signal sequence.
the 'synthetic' pathway • LE then move to the Golgi compartment to acquire mannose 6 -phosphate (M6 - P) ligand (marker) • Process requires the sequential action of two enzymes, • a phosphotransferase (Reitman & Kornfeld, 1981; Waheed et al, 1981) • a diesterase (Varki & Kornfeld, 1981; Waheed et al, 1981).
the 'synthetic' pathway • M6-P marker separates glycoproteins destined for the lysosome from secretory glycoproteins • Failure to acquire this marker results in mistargeting of lysosomal enzymes; they will not enter the lysosome and substrate breakdown will not occur.
Formation of lysosomal recognition tag or marker, mannose-6-phosphate
the 'synthetic' pathway • In the mucolipidoses II (I-cell disease) and III (pseudo-Hurler polydystrophy, mistargetting is precisely what happens • These patients lack the first enzyme, i.e. the phosphotransferase • Since all enzymes requiring the M6-P marker fail to enter the lysosome, they end up outside the cell • Consequently, these patients have very high plasma levels of all lysosomal enzymes • This clinical finding led to the discovery of the M6-P ligand and its receptor (Hickman & Neufeld, 1972).
the 'synthetic' pathway • Not all lysosomal enzymes require the M6-P ligand • Glucocerebrosidase (Gaucher disease) which is associated with the lysosomal membrane, does not acquire the M6-P residue, although it does undergo N-glycosylation and is targeted • The precise mechanism by which this occurs is unknown.
the 'synthetic' pathway • The receptor–protein complex then moves to the late endosome, where the low pH causes it to dissociate (Gonzalez-Noriega et al, 1980) • The hydrolase moves on into the lysosome and the receptor is recycled either to the Golgi to pick up another ligand, or to the plasma membrane. • The final steps in the maturation of the lysosomal enzyme include proteolysis, folding and aggregation.
Transport of macromolecules to the lysosome (the endocytotic pathway) • The material to be broken down in lysosomes may be extracellular or intracellular • Extracellular materials enter the cell either by endocytosis or phagocytosis, depending on the nature of the molecule. • Receptor-mediated endocytosis is the process by which most biologically important extracellular substances are internalized
the ‘endocytotic’ pathway • This occurs by binding to specific cell surface receptors (Goldstein et al, 1985) • Ligands are first delivered to early endosomes, and then transported to late endosomes, probably by multivesicular bodies • They are then delivered to the lysosomes.
the ‘endocytotic’ pathway • Phagocytosis is the route of entry into the cell for microorganisms and cellular debris • Such particles are incorporated into phagosomes, which fuse with primary lysosomes to form secondary lysosomes • Finally, intracellular materials undergo autophagy
the ‘endocytotic’ pathway • Although a small amount of hydrolysis takes place in endosomes, the bulk of it takes place in the lysosome • This is because it is only in the acid milieu of the lysosome that hydrolases are active • The low pH of the lysosome is maintained by the vacuolar proton pump
Retrograde transport from endosomes to the trans-Golgi network • Some intracellular transmembrane proteins, such as acid-hydrolase receptors, processing peptidases and SNAREs*, undergo retrograde transport from endosomes to the trans-Golgi network (TGN) as part of their normal trafficking • This retrograde-transport pathway is exploited by a subset of bacterial and plant protein toxins to enable them to reach the endoplasmic reticulum and eventually the cytosol. *Soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein receptors
Retrograde transport • Recent studies have begun to unravel the molecular machinery that is involved in this retrograde transport. • Acid-hydrolase receptors such as S. cerevisiae vacuolar sorting-10 protein (Vps10) and the mammalian mannose 6-phosphate receptors (MPRs) are selected for retrograde transport by a five-subunit complex named 'retromer' • In mammalian cells, this complex is mainly associated with tubules that emanate from vacuolar, early–late endosomal intermediates.
Retrograde transport • Although MPRs seem to be capable of being transported to the TGN from both early and late endosomal compartments, protein toxins undergo retrograde transport exclusively from early endosomes. • Not surprisingly then, protein toxins and MPRs share some components of the retrograde-transport machinery that is associated with early endosomal compartments
Retrograde transport • The location of molecular devices that are involved in the retrograde transport of MPRs and protein toxins highlights the existence of an extensive 'tubular endosomal network' (TEN) • This TEN sorts and recycles cargoes to various destinations, including different domains of the plasma membrane, the limiting membrane of lysosomes and lysosome-related organelles, and specialized storage vesicles, in addition to the TGN
Components of the molecular machinery that mediates retrograde transport from endosomes to the trans-Golgi network.
RETROMER COMPLEX • Multisubunit complex that mediates the retrograde transport of acid-hydrolase receptors between endosomes and the trans-Golgi network (TGN) • Recent studies have shown low level of retromer in affected regions of the brain of patients with Alzheimer disease.
PAUSE Working model of Retromer