Download

1 / 16
160 likes | 267 Views
EST Sequence Cleaning and Quality Control. FW4089 Bioinformatics H. Jiang. Background. Expressed Sequence Tag (EST): small pieces of DNA seq (300-500bp) sequenced at one or both ends of an expressed gene. Advantage: fast and easy, to fish a gene out.

E N D
EST Sequence Cleaning and Quality Control FW4089 Bioinformatics H. Jiang
Background • Expressed Sequence Tag (EST): small pieces of DNA seq (300-500bp) sequenced at one or both ends of an expressed gene. • Advantage: fast and easy, to fish a gene out. • Challenge: seq errors, redundant ESTs,.. • ~12, 000 from 8 cDNA libraries: • Control Apex (CA), Transgenic Apex (TA) • Control Apex (CL), Transgenic Apex (TL) • Control Apex (CR), Transgenic Apex (TR) • Control Apex (CS), Transgenic Apex (TS)
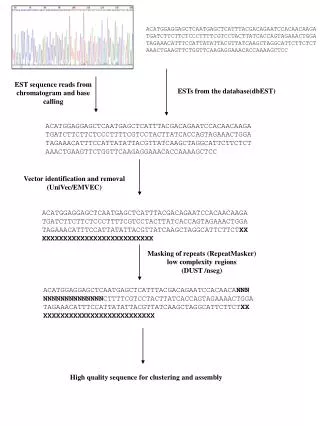
Sequence Cleaning (1) • Base-calling program---Phred: • Input: chromatogram files • Output: seq file and quality file (scores) >phred –id ./passTrace –sd ./passSeq/ -qd ./passQual/ • DNA sequence cleaning: quality trimming and vector removal---Lucy: • Lucy Steps: • Read input seq#, seq info, and quality info • Chop off splice sites • Remove vector insert • Produce output seq for fragment assembly.
Sequence Cleaning (2) • Lucy Input: seq file and quality file >lucy [parameters] seqFile qualFile [2ndSeqFile] • Lucy major parametersto set up: -vector vector_completeSeq splice_site_file (splice_site_file: 2 splice-site seq before and after the insertion point on the vector) splice.begin(150 bp) ____________________________________________________________________________ M13 Rev. Primer EcoRI Linker 1 Linker 4 EcoRI splice.end(150 bp) ____________________________________________________________________________ half of Linker 1 Linker 4 EcoRI M13 For Primer • Lucy Output: • identified locations of good/clean region • trim seq without vector, linker, Ns (<3% Ns), and poly A/Ts. • Other check-up (e.g. chloroplast, mitochondrial, …) • Submit 11, 545 ESTs to GenBank
Any Problem? An example. • In-house re-sequence clone at plate position of NP9A.A1, its seq should match MTU2CA.P16.F09, which was submitted. • However, blast ESTdb at NCBI did not find any match with our seq. • Problems? • Half of the new seq is vector contamination • Seq cleaning problems? • ClustalW-multiple seq alignment of new seq, Lucy-trimmed seq and Seq from seq company.
EcoRI linker seq NP_9A_A1_new_ AGAATACGCCAGCTTGGTACCGAGCTCGGATCCCTAGTAACGGCCGCCAG Lucy2CA.P16.F09_299bp_ -------------------------------------------------- Larkun-trimmed_751bp_ ---------------------------------CCTNGNACGGCCGC-AG NP_9A_A1_new_ TGTGCTGGAATTCGCCCTTTCGAGCGGCCGCCCGGGCAGGTCAACGGTTG Lucy2CA.P16.F09_299bp_ ----------------------------------------GCAACGGTTG Larkun-trimmed_751bp_ TGTGCTGGAATTCGCCCTTTCGAGCGGCCGCCCGGGCAGGNNAACGGTTG ******** NP_9A_A1_new_ ACTTCAATCCAACCATAATTAAACGGCGATAGATCATAATTTCAGTCAAG Lucy2CA.P16.F09_299bp_ ACTTCAATCCAACCATAATTAAACGGCGATAGATCATAATTTCAGTCAAG Larkun-trimmed_751bp_ ACTTCAATCCAACCATAATTAAACGGCGATAGATCATAATTTCAGTCAAG ************************************************** NP_9A_A1_new_ TTCTAAGAACCCATTATCAAATTATTATCCAACAACAACAATAATAATTT Lucy2CA.P16.F09_299bp_ TTCTAAGAACCCATTATCAAATTATTATCCAACAACAACAATAATAATTT Larkun-trimmed_751bp_ TTCTAAGAACCCATTATCAAATTATTATCCAACAACAACAATAATAATTT ************************************************** NP_9A_A1_new_ CTCATTCGAAGAGAATCGTAGAATTCATAATCTAAATCGAAAAAAAAACT Lucy2CA.P16.F09_299bp_ CTCATTCGAAGAGAATCGTAGAATTCATAATCTAAATCGAGGGGGAAACT Larkun-trimmed_751bp_ CTCATTCGAAGAGAATCGTAGAATTCATAATCTAAATCGNNNGGNNAACT *************************************** **** NP_9A_A1_new_ AAAAATCCATCAAATTAAACAAAAACAAACCCGAAGATGGATGAATCAAT Lucy2CA.P16.F09_299bp_ AAAAATCCATCAAATTAAACAAAAACAAACCCGAAGATGGATGAATCAAT Larkun-trimmed_751bp_ AAAAATCCATCAAATTAAACAAAAACAAACCCGAAGATGGATGAATCAAT ************************************************** NP_9A_A1_new_ TGGTCTTGGGATCAGGAAGGGGGAGAAAACCAGNACCAGGAGCAGAACCA Lucy2CA.P16.F09_299bp_ TGGTCTTGGGATCAGGAAGGGGGAGAAAACCAGCACCAGGAGCAGAACCA Larkun-trimmed_751bp_ TGGTCTTGGGATCAGGAAGGGGGAGAAAACCAGCACCAGGAGCAGAACCA ********************************* **************** NP_9A_A1_new_ CGAGGCGGAGAAAACCCAAGAAACTTCTGCAAATTAGTGCCTCGGCCGCG Lucy2CA.P16.F09_299bp_ CGAGGCGGAGAAAACCCAAGAAACTTCTGCAAATTAGTG----------- Larkun-trimmed_751bp_ CGAGGCGGAGAAAACCCAAGAAACTTCTGCAAATTAGTGCCTCGGCCGCG *************************************** NP_9A_A1_new_ ACCACGCTAAGGGCGAATTCTGCAGATATCCATCACACTGGCGGCCGCTC Lucy2CA.P16.F09_299bp_ -------------------------------------------------- Larkun-trimmed_751bp_ ACCACGCTAAGGGCGAATTCTGCAGATATCCATCACACTGGCGGCCGCTC Seqdifference
Find Questionable Sequences • Strategy: • Find and compare sequences of ‘clean region’ defined by Lucy (coordinates are in debug files produced by Lucy) • Find those seq that have low quality scores and different between Phred base calls and Lark calls • Criteria: minimum length >100, >10 continuous low quality scores (<16) • Compare difference between Lark and Lucy seq • Manually check some chromatogram files
Program 1 • From debug file get coordinate file: • Where good/clean region starts and ends • Program 1: (a C program) Usage: ./clean coordfile qualfile threshhold regionlength [outputfile] 1. get coordfile: ESTID+CLR 2. qualfile: quality file3. threshold: 16 (Lucy default)4. regionlength: 10 (10 continuous low scores) 5. optional output filename.
Program 2 Program 2: (a unix-based Flex program) Partial code: 1. Define rules (patterns): e.g., CHAR [A-Z]; INT -?[0-9]; MTUID; BAD {INT}* 2. Action {BAD} { if ( ? <=16 ) { ++count; } if (count >= 10 && inbadregion = 0) { strncp (?)? printf("\n%s ", yytext); } else { count = 0; inbadregion =1; } 3. main() 4. Compile and produce an executable file.
Result (1) • By C and Flex program, found ~2400 seq had 10 consecutive low scores • Lucy claimed that average probability of error (score<=16) over the final clean range is 2.5%. • Next: compare base calls between Lucy trimmed and Lark sequences. By 2 methods: GAP and Local Blast
GAP of 2400 seq pairs (1) • Convert file format: • gcg>fromfasta bigfastaFile of Lark and Lucy • Run a shell script: • gap Lucyseq1 Larkseq1 (all 2400 pairs) • Output: *.pair file (example see below) GAP of: mtu2ta.p13.h06.seq check: 4559 from: 1 to: 358 MTU2TA.P13.H06 to: origmtu2ta.p13.h06.seq check: 438 from: 1 to: 358 origMTU2TA.P13.H06 Gap Weight: 50 Average Match: 10.000 Length Weight: 3 Average Mismatch: 0.000 Quality: 3248 Length: 369 Ratio: 9.073 Gaps: 4 Percent Similarity: 99.712 Percent Identity: 97.983
GAP of 2400 seq pairs (2) • Perl program to parse *.pair output • Use regular expression to find key words and extract the corresponding values: if ($line=~/Ratio: /) {$line =~s/Gaps/:Gaps/;@crx =split (":",$line);$crx[1]=~s/^\s*//;$crx[1]=~s/\s*$//;$crx[3]=~s/^\s*//;$crx[3]=~s/\s*$//;$ratio =$crx[1];$gaps =$crx[3]; • Criteria: min length >100, gap>=1, identity <95%. • Found: total 188 seq different.
Local Blast of 2400 seq pairs (1) • Format Lark orig seq to database files >formatdb –i orig –p F –o T • Blast Lucy trimmed seq (one bigfastaFile) with orig >blastall –i lucy –d orig –p blastn –e 0.05 –m 9 –b 1 >blastout (output redireciton) • Blast output: # BLASTN 2.2.4 [Aug-26-2002] # Query: MTU2CA.P10.A07 # Database: orig # Fields: Query id, Subject id, % identity, alignmentlength, mismatches, gap openings, q. start, q. end, s. start, s. end, e-value, bit score MTU2CA.P10.A07 MTU2CA.P10.A07 99.83 573 1 0 1 573 1 573 0.0 1130.4 MTU2CA.P10.A07 MTU2CA.P4.E01 99.48 573 3 0 1 573 1 573 0.0 1114.6 MTU2CA.P10.A07 MTU2CA.P4.A02 99.07 540 5 0 34 573 6 545 0.0 1033.3
Local Blast of 2400 seq pairs (2) • Cut-off criteria: pick the top hit(same IDs), select those with identity <95%, alignmentlength>100, and gap>=1 • Parse the output by a Perl program • Found total 158 sequences.
Compare GAP and Blast Results • Compare results: Blast is quick and not accurate (e.g. unknown reason to open a gap for a perfect matched seq pair). • Find common seq IDs: • total of 144 seq (1.2% of 11, 545 submitted seq). • Manual check on their chromatograms (To be continued……)
A chromatogram example NNNGGNN