Download

1 / 24
240 likes | 420 Views
LECTURES 17-18 CATABOLISM OF PROTEINS AND AMINO ACID NITROGEN DR SAMEER FATANI BIOCHEMISTRY (METABOLISM). CATABOLISM OF PROTEINS AND AMINO ACIDS Molecular Nitrogen (N2) is available in atmosphere, and before

E N D
LECTURES 17-18 CATABOLISM OF PROTEINS AND AMINO ACID NITROGEN DR SAMEER FATANI BIOCHEMISTRY (METABOLISM)
CATABOLISM OF PROTEINS AND AMINO ACIDS Molecular Nitrogen (N2) is available in atmosphere, and before It can be utilized by animals, it must be reduced (fixed) from N2 to NH3 by microorganisms, and plants. Ammonia is then incorporated into amino acids and proteins. these proteins finally become part of the food chain. Humans can synthesize only 11 of the 20 amino acids (non- essential). The amino acids that can not be synthesized by humans “de Novo” are termed “essential” because they must be obtained from dietary foodstuffs that contain them. Products of amino acid metabolism “such as NADH” can be used to provide energy.
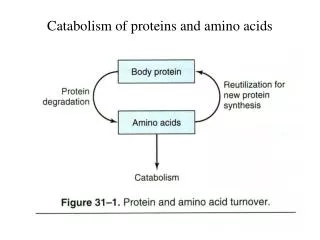
Nitrogen balance A healthy adult human that eat varied and plentiful diet Is generally in “nitrogen balance”. What is nitrogen balance? A state where the amount of nitrogen ingested each day Is balanced by the amount excreted. (no change in the amount of body Nitrogen). What is protein turnover? Protein turnover is defined as the synthesis and Degradation of protein. The body is either in negative or positive nitrogen balance: In negative nitrogen balance: more nitrogen is excreted than ingested. This occurs in starvation and certain diseases: in starvation Carbon chains of amino acids from proteins are needed for Gluconeogenesis; ammonia released from amino acids is excreted as urea and is not incorporated into protein.
Positive nitrogen balance: more ingestion of nitrogen and less excretion. occurs in growing children, who are increasing their body weight and incorporating more amino acids into proteins than they breakdown. Positive nitrogen balance also occurs in pregnancy and during refeeding after starvation.
MECHANISM OF ACTION OF AMINOTRANSFERASES • Aminotransferases used: • to transfer amino group from one amino acid to another • or to transfer amino group for degradation of certain amino acid. (fig.18.3—D5) • Nonessential amino acids are synthesized from α-keto acid precursors via transfer of preexisting amino group from another amino acid by aminotransferases (transaminase). (fig.18.4—D5). • Transamination involving essential amino acids is normally unidirectional because the body can not synthesize the equivalent α-keto acid. (fig.18.5—D5). • Only threonine and lysine amino acids do not participate in • Aminotransferase reaction. • Aminotransferase reactions are reversable except for essential A.A. • The α-keto acids are transaminated by aminotransferases to produce the different amino acid.
Pyridoxal phosphate as cofactor for aminotransferase Transfer of amino group occurs in the presence of the enzyme Aminotransferases and the cofactor pyridoxal phosphate. The active site of the amiotransferase contains pyridoxal Phosphate covalently attached to the amino group of lysine residue in aminotransferse. (fig.18.8—D5). The covalent linkage which formed between the aminotransferase And the cofactor (pyridoxal phosphate) termed “Schiff base” (-CH=N-). (fig.18.8). C= originates in the aldehyde group of pyridoxal pho. N= donated by lysine residue. During degradation of amino acids (A A): -A A approaches the active site of aminotransferase -The amino group of A A displace the lysine amino group and form “Schiff base” linkage with pyridoxal pho. - Hydrolysis of A A will give : α-keto acid and pyridox-amine
A.A. to be degraded Aminotransferase enzyme 1 A.A. approaches Schiff base lysine Schiff base (-CH=N-) 2 aldehyde Amino group of A.A. displace Lysine amino group of the enzyme Pyridoxal phosphate cofactor 3 Amino group of A.A. Form Schiff base With pyri. phosphate 4 Hydrolysis of A. A. give: α-keto acid and pyridox amine
Glutamate dehydrogenase incorporate and produces ammonia Ammonia in liver incorporated into glutamate by glutamate Dehydrogenase. The enzyme also catalyzes the reverse reaction. (fig. 18.11—D5) The enzyme produces ammonia from amino acids when these amino acids are needed as glucose precursors or for energy. A major source of ammonia is bacterial metabolism in the intestinal lumen, the released ammonia will be absorbed and transported to the liver. Glutamate dehydrogenase incorporates this ammonia into Glutamate. Regulation of glutamate dehydrogenase Glutamate dehydrogenase is regulated allosterically by purine nucleotide (18.13—D5).
Toxicity and transport of ammonia Ammonia transporter (glutamine) Free ammonia is toxic and is preferentially transported in the blood In the form of amino or amide groups. 50% of circulating amino acid molecules areglutamine “the ammonia Transporter”. The amide group of glutamine is a nitrogen donor for several classes of Molecules including: purine bases, and the amino group of cystine. Glutamate and ammonia are the substrates for glutamine synthetase. (18.14—D5). Removal of the amide group is catalyzed by glutaminase (18.15---D5).
Amino acid oxidase The function: removal of amino groups from amino acids. The substrate: amino acid The final products: α-keto acid, ammonia, and water, “the same Products as for glutamate dehydrogenase reaction”. In amino acid oxidase reaction, unlike the reaction catalyzed by Glutamate dehydrogenase, there is noconcomitant production Of NADH and therefore no ATP production. (18.19– D5)
TRANSPORT OF NITROGEN TO LIVER AND KIDNEY • Amino acids are transported from muscle after proteolysis • - The most percentage of body protein is in skeletal muscle. • - Under conditions of energy need, this protein is degraded and amino • groups from amino acids are transferred to produce glutamine and • alanine. • Finally, these amino acids “glutamine and alanine” are transported to • Liver and kidney to produce urea and ammonia. • Muscle protein responds to conditions such as: starvation, trauma, • Burns, and septicemia by undergoing massive degradation. • Ammonia is released in liver and kideny • The main destination of glutamine and alanine in the blood is the liver, • where ammonia is released by alanine aminotransferase, glutaminase, • And glutamate dehydrogenase. • Glutamate dehydrogenase not only releases ammonia but also produces • NADH and α-keto-glutarate as source of energy. • Cachexia: a condition produced by many malignancies, characterized • By increasing the rate of amino acid removal from plasma by the liver, • Subsequently activating muscle proteolysis. • Some glutamine and alanine is taken up by the kidneys. Ammonia is • Released by the same enzymes that are active in liver, ammonia will be • Protonated to ammonium and excreted.
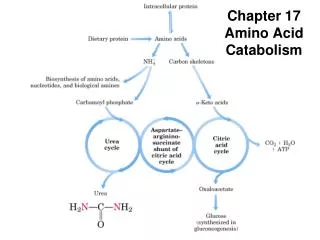
UREA CYCLE The urea cycle is the major mechanism for utilization and excretion Of ammonia (excretion of ammonia in form of urea in the kidneys). The precursors of urea cycle: ammonium ion and bicarbonate, Both will form carbamoyl phosphate. Carbamoyl phosphate and citrulline, the first two intermediates of urea cycle are synthesized in mitochondria, while the rest of the Reactions in the cytosol. In urea cycle: urea will be produced, since humans cannot metabolize Urea it is transported to the kidneys for filtration and excretion. SEE THE NEXT DIAGRAM FOR THE WHOLE STEPS OF UREA CYCLE
UREA CYCLE HCO3- + NH4+ 2ATP Carbamoyl phosphate synthetase 2ADP + Pi Carbamoyl phosphate Ornithinetranscarbamoylase UREA Aspartate ATP Argininosuccinate synthetase Arginase PPi + AMP Argininosuccinate lyase Fumarate
METABOLIC DISORDERS OF UREA SYNTHESIS Ammonia is a very toxic substance, and usually excreted in the form of urea. Metabolic disorders that arise from abnormal function of enzymes Of urea synthesis (urea cycle) are fatal and cause coma “due to ATP depletion”, specially when ammonia concentration is high. High concentration of ammonia sequesters α-ketoglutarate in form of Glutamate, leading to: - depletion of TCA cycle intermediates - and reducing ATP production Therapy for the deficiencies in urea cycle enzymes depend on The following: 1- to limit protein intake and build up of ammonia. 2- to remove excess ammonia 3- To replace any intermediates missing from urea cycle.
Deficiencies of urea cycle enzymes Ornithine transcarbamoylase Deficiency : The cause: lack of transcarbamoylase The results: Mental retardation, and death. Therapy: adequate therapy could be useful. Mental retardation might be caused by the excess ammonia. The gene for ornithine transcarbamoylase is on the X chromosome, And males are generally more affected seriously. Argininosuccinate synthetse deficiency: The cause: the inability to condense citrulline with aspartate “to form Argininosuccinate”. The results: accumulation of citrulline in blood and its excretion In urine (citrulinemia). Therapy: requires specific supplementation with arginine for protein Synthesis. Arginase deficiency: The cause: lack or arginase The results: causes many abnormalities in development and function of the central nervous system. Arginine accumulates and is excreted. Therapy: A diet including essential amino acids but excluding arginine has been used effectively.
METABOLIC DISORDERS OF UREA SYNTHESIS Ammonia is a very toxic substance, and usually excreted in the form of urea. Metabolic disorders that arise from abnormal function of enzymes Of urea synthesis (urea cycle) are fatal and cause coma “due to ATP depletion”, specially when ammonia concentration is high. High concentration of ammonia sequesters α-ketoglutarate in form of Glutamate, leading to: - depletion of TCA cycle intermediates - and reducing ATP production Therapy for the deficiencies in urea cycle enzymes depend on The following: 1- to limit protein intake and build up of ammonia. By limiting the ingestion of amino acids, maybe replacing them if necessary with equivalent α-keto acids. By decreasing the bacterial source of ammonia in intestines, by using a compound that acidifies the colon (metabolized by colonic bacteria to acidic products) this promotes the excretion of ammonia in feces as protonated ammonium ions. Antibiotics can be also used to kill ammonia-producing bacteria.
2- to remove excess ammonia By compounds that bind covalently to amino acids and produce nitrogen-containing molecules that are excreted in urine. 3- To replace any intermediates missing from urea cycle. Deficiencies of urea cycle enzymes Ornithine transcarbamoylase Deficiency The cause: lack of transcarbamoylase The results: Mental retardation, and death. Therapy: adequate therapy could be useful. Mental retardation might be caused by the excess ammonia. The gene for ornithine transcarbamoylase is on the X chromosome, And males are generally more affected seriously. Argininosuccinate synthetse deficiency: The cause: the inability to condense citrulline with aspartate “to form Argininosuccinate”. The results: accumulation of citrulline in blood and its excretion In urine (citrulinemia). Therapy: requires specific supplementation with arginine for protein Synthesis.