Download
1 / 58
580 likes | 596 Views
Explore the rationale and methods for understanding protein structure and function, including prediction through computational models. Learn about the complexity of protein folding, from primary to quaternary structures, through X-ray crystallography and NMR spectroscopy. Discover the significance of protein structure in rational drug design, genetic engineering, and cellular pathway modeling.

E N D
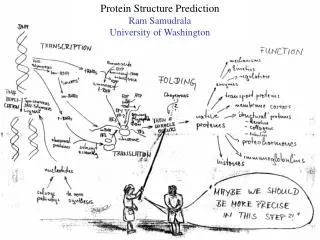
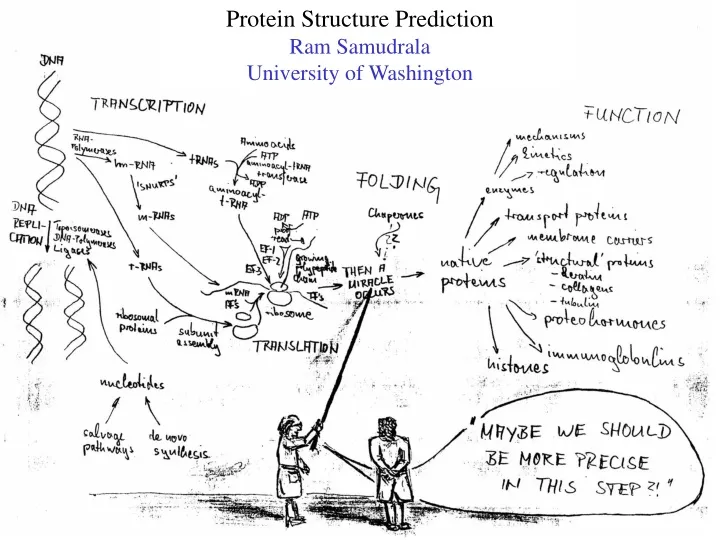
Protein Structure Prediction Ram Samudrala University of Washington
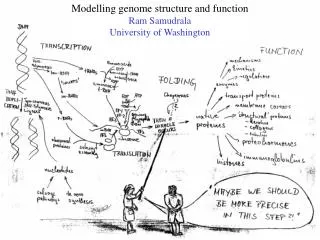
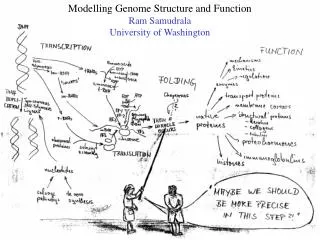
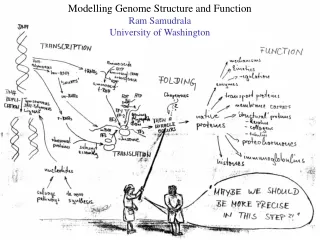
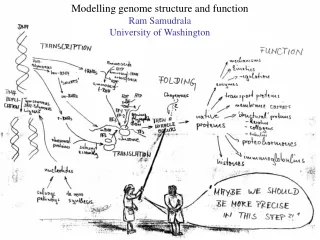
Rationale for understanding protein structure and function structure determination structure prediction Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) unique shape precisely ordered stable/functional globular/compact helices and sheets native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
Protein folding landscape Large multi-dimensional space of changing conformations J=10-3 s unfolded barrier height molten globule DG* free energy native J=10-8 s folding reaction
Protein primary structure twenty types of amino acids two amino acids join by forming a peptide bond R R H O H C H H N OH Cα Cα Cα OH N C N C H H O O H H R each residue in the amino acid main chain has two degrees of freedom (f and y) R R H O H O H H c c y f y f C N C f N f Cα Cα Cα Cα N C N C y y c c H H O H O H R R the amino acid side chains can have up to four degrees of freedom (c1-4)
Protein secondary structure many f,y combinations are not possible b sheet (anti-parallel) +180 b L a f 0 C -180 -180 0 y +180 N a helix C b sheet (parallel) N
Protein tertiary and quaternary structures Ribonuclease inhibitor (2bnh) Haemoglobin (1hbh) Hemagglutinin (1hgd)
Methods for determining protein structure X-ray crystallography NMR spectroscopy Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
X-ray crystallography- concept • X-rays interact with electrons in protein molecules arranged in a crystal to produce • diffraction patterns • The diffraction patterns of the x-rays can be used to determine the three-dimensional • structure of proteins • Provides a “static” picture From <http://info.bio.cmu.edu/courses/03231/LecF01/Lec25/lec25.html>
X-ray crystallography- details • Prepare protein crystals where the proteins are organised in a precisecrystal lattice • Shine x-rays on crystals which diffract off of electrons of atoms inthe crystals; the • intensities of the individual reflections aremeasured • Phases are usually obtained indirectly by ismorphous replacement, fromthe way one or • a few heavy atoms incorporated into the sameisomorphous crystal lattice affect the • diffraction patern • Intensities and phases of all reflections are combined in a Fouriertransform to provide • maps of electron density • Interpret the map by fitting the polypeptide chain to the contours • Refine the model by minimising the distance between the observedamplitudes and the • calculated amplitudes
NMR spectroscopy - concept NK-lysin (1nkl) S1 RNA binding domain (1sro) • The magnetic-spin properties of atomic nuclei within a molecule areused to obtain a • list of distance constraints between atoms in themolecule, from which a • three-dimensional structure of the proteinmolecule can be obtained • Provides a “dynamic” picture
NMR spectroscopy - details • Protein molecules placed in a strong magnetic field have theirhydrogen atoms aligned • to the field; the alignment can be excited byapplying radio frequency (RF) pulses • Possible to obtain unique signal (chemical shift) for each hydrogenatom in a protein • molecule • Structural information arises primarily from the Nuclear OverhauserEffect (NOE), • which gives information about distances between atoms ina molecule • A pair of protons give a detectable NOE cross-peak if they are within5.0 Åof each • other in space • After obtaining NOE data for protons througout the structure, a numberof independent • structures can be generated that are consistent withthe distance constraints
Computer representation of protein structure • Structures are stored in the protein data bank (PDB), arepository of mostly • experimental models based on X-raycrystallographic and NMR studies • <http://www.rcsb.org> • Atoms are defined by their Cartesian coordinates: • ATOM 1 N GLU 1 18.222 18.496 -16.203 1.00 21.95 • ATOM 2 CA GLU 1 17.706 17.982 -14.905 1.00 16.74 • ATOM 3 C GLU 1 17.368 16.466 -15.121 1.00 15.45 • ATOM 4 O GLU 1 16.780 16.073 -16.175 1.00 18.81 • ATOM 5 CB GLU 1 16.552 18.744 -14.351 1.00 17.35 • ATOM 6 CG GLU 1 16.952 20.118 -13.803 1.00 24.48 • ATOM 7 CD GLU 1 15.881 21.145 -13.597 1.00 31.51 • ATOM 8 OE1 GLU 1 16.012 22.316 -13.292 1.00 29.12 • ATOM 9 OE2 GLU 1 14.701 20.768 -13.799 1.00 35.19 • ATOM 10 N PHE 2 17.762 15.746 -14.052 1.00 15.83 • ATOM 11 CA PHE 2 17.509 14.262 -14.184 1.00 13.24 • These structures provide the basis for most of theoretical workin protein folding and • protein structure prediction
Comparison of protein structures 3.6 Å 2.9 Å NK-lysin (1nkl) Bacteriocin T102/as48 (1e68) T102 best model • Need ways to determine if two protein structures are related and to compare predicted • models to experimental structures • Commonly used measure is the root mean square deviation (RMSD)of the Cartesian • atoms between two structures after optimalsuperposition (McLachlan, 1979): • Usually use Caatoms • Other measures include contact maps and torsion angle RMSDs
Methods for predicting protein structure comparative modelling fold recognition ab initio prediction Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
Comparative modelling of protein structure • Proteins that have similar sequences (i.e., related by evolution)have similar • three-dimensional structures • A model of a protein whose structure is not known can be constructedif the structure of • a related protein has been determined byexperimental methods • Similarity must be obvious and significant for good models to be built • Need ways to build regions that are not similar between the tworelated proteins • Need ways to move model closer to the native structure
Comparative modelling of protein structure scan align KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … build initial model construct non-conserved side chains and main chains refine
Fold recognition 3.6 Å 5% ID NK-lysin (1nkl) Bacteriocin T102/as48 (1e68) • The number of possible protein structures/folds is limited (largenumber of sequences • but few folds) • Proteins that do not have similar sequences sometimes have similarthree-dimensional • structures • A sequence whose structure is not known is fitted directly (or“threaded”) onto a known • structure and the “goodness of fit” isevaluated using a discriminatory function • Need ways to move model closer to the native structure
Fold recognition evaluate fit KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … build initial model construct non-conserved side chains and main chains refine
Ab initio prediction of protein structure – concept • Go from sequence to structure by sampling the conformational space ina reasonable • manner and select a native-like conformation using a gooddiscrimination function • Problems: conformational space is astronomical, and it is hard todesign functions that • are not fooled by non-native conformations (or“decoys”)
Ab initio prediction of protein structure select sample conformational space such that native-like conformations are found hard to design functions that are not fooled by non-native conformations (“decoys”) astronomically large number of conformations 5 states/100 residues = 5100 = 1070
Sampling conformational space – continuous approaches energy • Most work in the field • Molecular dynamics • Continuous energy minimisation (follow a valley) • Monte Carlo simulation • Genetic Algorithms • Like real polypeptide folding process • Cannot be sure if native-like conformations are sampled
Molecular dynamics • Force = -dU/dx (slope of potential U); acceleration, m a(t) = force • All atoms are moving so forces between atoms are complicated functions of time • Analytical solution for x(t) and v(t) is impossible; numerical solution is trivial • Atoms move for very short times of 10-15 seconds or 0.001 picoseconds (ps) • x(t+Dt) = x(t) + v(t)Dt + [4a(t) – a(t-Dt)] Dt2/6 • v(t+Dt) = v(t) + [2a(t+Dt)+5a(t)-a(t-Dt)] Dt/6 • Ukinetic = ½ Σ mivi(t)2 = ½ n KBT • Total energy (Upotential + Ukinetic) must not change with time old position old velocity acceleration new position old velocity acceleration new velocity n is number of coordinates (not atoms)
Energy minimisation starting conformation energy deep minimum number of steps energy give up steepest descent conjugate gradient number of steps converge RMSD • For a given protein, the energy depends on thousands of x,y,z Cartesian atomic • coordinates; reaching a deep minimum is not trivial • With convergence, we have an accurate equilibrium conformation and a well-defined • energy value
Monte Carlo simulation • Discrete moves in torsion or cartesian conformational space • Evaluate energy after every move and compare to previous energy (DE) • Accept conformation based on Boltzmann probability: • Many variations, including simulated annealing (starting with ahigh temperature so • more moves are accepted initially and thencooling) • If run for infinite time, simulation will produce a Boltzmmandistribution
Genetic Algorithms • Generate an initial pool of conformations • Perform crossover and mutation operations on this set to generatea much larger pool of • conformations • Select a subset of the fittest conformations from this large pool • Repeat above two steps until convergence
Sampling conformational space – exhaustive approaches select enumerate all possible conformations view entire space (perfect partition function) must use discrete state models to minimise number of conformations explored computationally intractable: 5 states/100 residues = 5100 = 1070 possible conformations
Scoring/energy functions • Need a way to select native-like conformations from non-native ones • Physics-based functions: electrostatics, van der Waals, solvation, bond/angle terms • Knowledge-based scoring functions: derive information about atomic properties from a • database of experimentally determined conformations; common parametres include • pairwise atomic distances and amino acid burial/exposure.
Requirements for sampling methods and scoring functions • Sampling methods must produce good decoy sets that are comprehensive and include • several native-like structures • Scoring function scores must correlate well with RMSD of conformations (the better • the score/energy, the lower the RMSD)
Overview of CASP experiment • Three categories: comparative/homology modelling, foldrecognition/threading, and • ab initio prediction • Goal is to assess structure prediction methods in a blind andrigourous manner; blind • prediction is necessary for accurateassessment of methods • Ask modellers to build models of structures as they are in the processof being solved • experimentally • After prediction season is over, compare predicted models to theexperimental • structures • Discuss what went right, what went wrong, and why • Compare progress from CASP1 to CASP4 • Results published in special issues of Proteins: Structure, Function, Genetics 1995, • 1997, 1999, 2002
Comparative modelling at CASP - methods • Alignment: PSI-BLAST, FASTA, CLUSTALW - multiple sequencealignments • carefully hand-edited using secondary structure information • More successful side chain prediction methods include: • backbone-dependent rotamer libraries (Bower & Dunbrack) • segment matching followed by energy minimisation (Levitt) • self-consistent mean field optimisation (Bates et al) • graph-theory+ knowledge-basedfunctions (Samudrala et al) • More successful loop building methods include: • satisfaction of spatial restraints (Sali) • internal coordinate mechanics energy optimisation (Abagyan et al) • graph-theory + knowledge-basedfunctions (Samudrala et al) • Overall model building: there is no substitute for careful hand-constructed models • (Sternberg et al, Venclovas)
A graph theoretic representation of protein structure -0.6 (V1) represent residues as nodes -0.5 (I) -0.9 (V2) weigh nodes -0.7 (K) -1.0 (F) construct graph -0.6 (V1) -0.2 -0.5 (I) -0.9 (V2) -0.1 -0.5 (I) -0.9 (V2) -0.1 -0.1 -0.3 -0.1 find cliques -0.2 -0.4 -0.3 -0.1 -0.1 -0.4 W = -4.5 -0.2 -0.7 (K) -1.0 (F) -0.2 -0.7 (K) -1.0 (F)
Historical perspective on comparative modelling alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å
Historical perspective on comparative modelling alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å
Prediction for CASP4 target T128/sodm Ca RMSD of 1.0 Å for 198 residues (PID 50%)
Prediction for CASP4 target T111/eno Ca RMSD of 1.7 Å for 430 residues (PID 51%)
Prediction for CASP4 target T122/trpa Ca RMSD of 2.9 Å for 241 residues (PID 33%)
Prediction for CASP4 target T125/sp18 Ca RMSD of 4.4 Å for 137 residues (PID 24%)
Prediction for CASP4 target T112/dhso Ca RMSD of 4.9 Å for 348 residues (PID 24%)
Prediction for CASP4 target T92/yeco Ca RMSD of 5.6 Å for 104 residues (PID 12%)
Comparative modelling at CASP - conclusions alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å CASP2 fair ~ 75% ~ 1.0 Å ~ 3.0 Å CASP3 fair ~75% ~ 1.0 Å ~ 2.5 Å CASP4 fair ~75% ~ 1.0 Å ~ 2.0 Å CASP4: overall model accuracy ranging from 1 Å to 6 Å for 50-10% sequence identity **T111/eno – 1.7 Å (430 residues; 51%) **T122/trpa – 2.9 Å (241 residues; 33%) **T128/sodm – 1.0 Å (198 residues; 50%) **T125/sp18 – 4.4 Å (137 residues; 24%) **T112/dhso – 4.9 Å (348 residues; 24%) **T92/yeco – 5.6 Å (104 residues; 12%)
Fold recognition at CASP - methods • Visual inspection with sequence comparison (Murzin group) • Procyon - potential of mean force based on pairwise interactionsand global dynamic • programming (Sippl group) • Threader - potential of mean force and double dynamic programming (Jones group) • Environmental 3D Profiles (Eisenberg group) • NCBI Threading Program using contact potentials and models ofsequence-structure • conservation (Bryant group) • Hidden Markov Models (Karplus group) • Combination of threading with ab initio approaches (Friesner group) • Environment-specific substitution tables and structure-dependent gap penalties • (Blundell group)
Fold recognition at CASP - conclusions • Fold recognition is one of the more successful approaches atpredicting structure at all • four CASPs • At CASP2 and CASP4, one of the best methods was simple sequence searching with • carefulmanual inspection (Murzin group) • At CASP3 and CASP4, none of the threading targets could have been recognised bythe • best standard sequence comparison methods such asPSI-BLAST • For the most difficult targets, the methods were able to predict 60 residuesto 6.0Å • Ca RMSD, approaching comparative modelling accuracies as the similarity between • proteins increased.
Ab initio prediction at CASP – methods • Assembly of fragments with simulated annealing (Simons et al) • Exhaustive sampling and pruning using knowledge-based scoringfunctions • (Samudrala et al) • Constraint-based Monte Carlo optimisation (Skolnick et al) • Thermodynamic model for secondary structure prediction withmanual docking of • secondary structure elements and minimisation(Lomize et al) • Minimisation of a physical potential energy function with asimplified representation • (Scheraga et al, Osguthorpe et al) • Neural networks to predict secondary structure (Jones, Rost)
Semi-exhaustive segment-based folding fragments from database 14-state f,y model generate … … monte carlo with simulated annealing conformational space annealing, GA minimise … … all-atom pairwise interactions, bad contacts compactness, secondary structure filter EFDVILKAAGANKVAVIKAVRGATGLGLKEAKDLVESAPAALKEGVSKDDAEALKKALEEAGAEVEVK
Historical perspective on ab initio prediction Before CASP (BC): “solved” (biased results) CASP2: worse than random with one exception CASP1: worse than random CASP3: consistently predicted correct topology - ~ 6.0 Å for 60+ residues *T56/dnab – 6.8 Å (60 residues; 67-126) **T61/hdea – 7.4 Å (66 residues; 9-74) **T64/sinr – 4.8 Å (68 residues; 1-68) **T75/ets1 – 7.7 Å (77 residues; 55-131) *T74/eps15 – 7.0 Å (60 residues; 154-213) **T59/smd3 – 6.8 Å (46 residues; 30-75) CASP4: ?
Prediction for CASP4 target T110/rbfa Ca RMSD of 4.0 Å for 80 residues (1-80)
Prediction for CASP4 target T97/er29 Ca RMSD of 6.2 Å for 80 residues (18-97)
Prediction for CASP4 target T106/sfrp3 Ca RMSD of 6.2 Å for 70 residues (6-75)