Download

1 / 26
270 likes | 534 Views
Progress in Calculating the Potential Energy Surface of H 3 + . Ludwik Adamowicz and Michele Pavanello , Department of Chemistry and Biochemistry. February 9th, 2012. UA SUPERCOMPUTERS Shared Memory - SGI Altix 4700

E N D
Progress in Calculating the Potential Energy Surface of H3+ LudwikAdamowicz and Michele Pavanello, Department of Chemistry and Biochemistry February 9th, 2012
UA SUPERCOMPUTERS • Shared Memory - SGI Altix 4700 • Marin - Interactive Front End, Altix 4700, 100-core Itanium2, 160 GB memory • Bora - Batch System, Altix 4700, • 512-core Itanium2, 1024 GB memory • Cluster - SGI Altix ICE 8200 • Ice - Interactive Front End, 3 "round-robin" login nodes cluster • 1392-core, quad-core Xeon (Harpertown), 2 GB memory/core • New Cluster - SGI Altix ICE 8400 • 2112-core, 6-core INTEL Xeon • Soon To Be Installed • SGI Altix UV • IBM: DataPlex HTC
Why Molecules with Hydrogen? • Goal: Accurate PES of H3+ • Motivation • Interstellar chemistry: (Hn)+ • Spectroscopy: H3+ • What has been done in the past? *CencekJCP, 108, 2831 (1998) ; Explicitly Correlated Gaussians (ECGs)
What are ECGs? • Expansion in terms of basis functions • The basis set is made of explicitly correlated Gaussians with floating centers Atoms Molecules The case of H3+ linear and non-linear parameters
The cusp condition The derivative of Ψ in this point counts! Electron-Nucleous cusp Electron-Electron cusp Kato’s condition* Cusp function: Is the nucleous really a point charge? *T. Kato (1957)
Optimization of ФM The step size is determined as a function of G and E.
Does our approach work?Single point calculation • Variational Principle Non-linearity: M*7 parameters Encounter linear dependencies 1) Pavanello et. al. J. Chem. Phys., 130, 034104 (2009) 2) Cencek et al. Chem. Phys. Lett. 246, 417-420 (1995)
Limits • 5 or 6 electrons maximum • Antisymmetrize electrons: ne! • Basis set size: M2 • Schrödinger Equation • Born-Oppenheimer approximation • Relativity • Coulomb Hamiltonian • Implementation – Parallelization – Numerical Instability • Encounter linear dependencies • Memory constraints
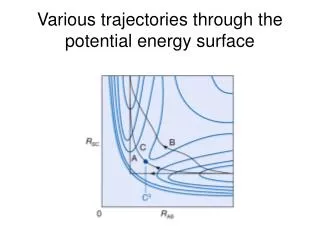
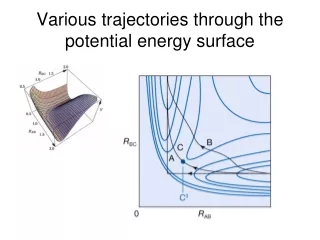
What if we move the geometry?Can we carry out PES calculations? • Re-optimize from scratch the basis set for each PES grid point. • Takes a long time to optimize the basis set • Hundreds, sometimes thousands of geometries need to be considered • Guess the basis set from nearby geometries • How? • Is it precise? • Is the precision maintained for each grid point? We need a benchmark!
Test of the spring method Benchmark PES of H3+ Generated a 377-point PES The wavefunction at a certain geometry was generated from one of a nearby geometry The spring model M=900 6300 parameters Convergence dictated by the value of the analytical gradient ( GTG < 10-11a.u. ) and not of the energy Pavanello et al. J. Chem. Phys. 130, 001033 (2009)
1st benchmark: D3h symmetry • We notice: • Our energies are always 0.01 cm-1 below the best in the literature. • Stretched geometries seem to show better improvement The more negative the better! ΔE(a.u. x 10-8) Total Energy (a.u.)
2nd benchmark: C2v & asymmetric asymmetric C2v 4 C2v 3
The challenge: a complete PES of H3+toward sub 0.01 cm-1 accuracy R13 R23 ρ (a.u.) R12 Total Energy (a.u.) Viegas, Alijah and Varandas, JCP (2007) Johnson, JCP (1980) Whitten and Smith (1968) Alijah at al. used MR-CI with cc-pV5Z
MR-CI vs "exact" H3+ [H H H]+ 2H+H+ Alijah’s most diffuse function Alijah et al. Our work (ECGs) Energy Difference (a.u.) 20 cm-1
Conclusions on H3+ • We developed: • ECG with analytical gradients, tested on single point calculations • Spring method to calculate PESs, tested on a 69 point PES portion of H3+ • The code is applicable to any (ne<7) molecular system • We achieved: • Most accurate variational energies to date • Most accurate (≈ 0.01cm-1) and extensive PES (42000 grid points) of H3+
Conclusions on H3+ • To be developed: • Leading relativistic corrections • Non-adiabatic corrections • Leading QED corrections
Equivalent treatment of nuclei and electrons in H3+ The total laboratory-frame nonrelativistic Hamiltonian:
Explicitly Correlated Gaussian Functions for non-BO calculations of H3+ diatomics H3+ or
Expectation values of the ground state non-BO energies, virial coefficients (η) , and internuclear distances for some isotopologues of H3+ . All values are calculated for an optimized 50 term explicitly correlated Gaussian basis set and are in atomic units.
Acknowledgements Coworkers: Pawel Kozlowski Donald Kinghorn Mauricio CafieroSergiyBubin Michele Pavanello Wei-Cheng Tung Collaborators: Alexander Alijah Nikolai Zobov Irina I. Mizus Oleg Polyansky Jonathan Tennyson TamásSzidarovzsky Attila Császár Max Berg AnnemiekePetrignani Andreas Wolf Support: NSF











![6.3 – Calculating [H 3 O + ] and [OH - ]](https://cdn3.slideserve.com/6515906/6-3-calculating-h-3-o-and-oh-dt.jpg)