Download
1 / 24
740 likes | 2.13k Views
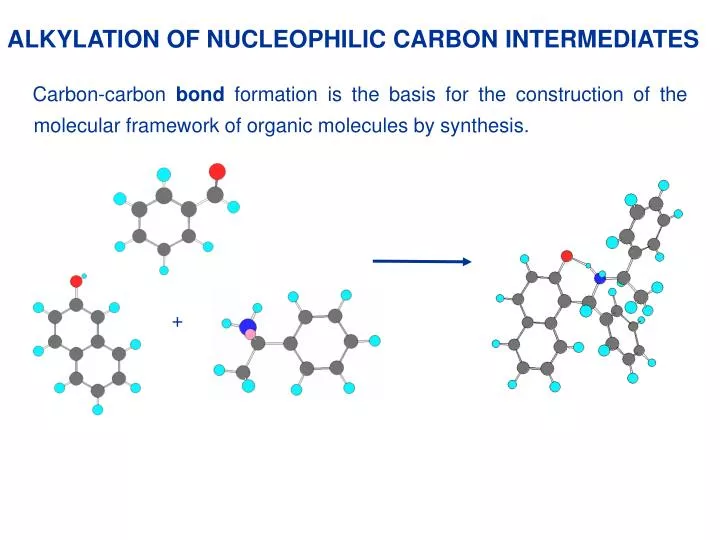
+. ALKYLATION OF NUCLEOPHILIC CARBON INTERMEDIATES. Carbon-carbon bond formation is the basis for the construction of the molecular framework of organic molecules by synthesis.

E N D
+ ALKYLATION OF NUCLEOPHILIC CARBON INTERMEDIATES Carbon-carbon bond formation is the basis for the construction of the molecular framework of organic molecules by synthesis.
One of the fundamental processes for carbon-carbon bond formation is a reaction between a nucleophilic carbon and an electrophilic one. enolate ions reactions with alkylating agents imine anions CARBON NUCLEOPHILES enamines SN2 mechanism The focus in this lesson is on enolate ions, imine anions and enamines, which are the most useful kinds of carbon nucleophiles, and on their reactions with alkylating agents. Mechanistically, these are usually SN2 reactions in which the carbon nucleophile displaces a halide or other leaving group.
SUCCESSFUL C-C BOND FORMATION Conditions for generation of the carbon nucleophile influenced by requires role of solvents, counterions and other components of the reaction media influenced by influenced by influenced by regio- and stereoselectivity of the alkylation reaction effect of the reaction conditions on the structure and reactivity of the nucleophile Successful carbon-carbon bond formation requires that the SN2 alkylation be the dominant reaction. SN2 ALKYLATION DOMINANT REACTION
1.1. GENERATION OF CARBANIONS BY DEPROTONATION • A very important means of generating carbon nucleophiles involves removal of a proton from a carbon by a Brønsted base. The anions produced are carbanions. • Both the rate of deprotonation and the stability of the resulting carbanion are enhanced by the presence of substituent groups that can stabilize negative charge. • A carbonyl group bonded directly to the anionic carbon can delocalize the negative charge by resonance, and carbonyl compounds are especially important in carbanion chemistry. • The anions formed by deprotonation of the carbon alpha to a carbonyl group bear most of their negative charge on oxygen and are referred to as enolates.
removal of a proton from a carbon by a Brønsted base enhance stability (thermodinamic) generates enhance carbon nucleophiles rate of deprotonation (kinetic) of of called generated on carbon atoms bonded directly to the carbonyl groups bear most of their negative charge on oxygen are referred to as enolates substituent groups that can stabilize negative charge carbanions
The efficient generation of a significant equilibrium concentration of a carbanion requires choice of a proper Brønsted base. • The equilibrium will favor carbanion formation only when the acidity of the carbon acid is greater than that of the conjugate acid corresponding to the base used for deprotonation. • Acidity is quantitatively expressed as pKa, which is equal to –log Ka, and applies, by definition, to dilute aqueous solution. Because most important carbon acids are quite weak acids (pKa > 15), accurate measurement of their acidity in aqueous solutions is impossible, and acidities are determined in organic solvents and referred to the pKa in an approximate way. The data produced are not true pKas, and their approximate nature is indicated by referring to them as simply pK values. • Tabulated pK values can indicate an order of some important substituents with respect to their ability to stabilize carbanions.
The position of the acid-base equilibrium for a given reactant-base combination can be estimated by comparing the approximate pK values of the conjugate acids of the bases with those of the carbon acid of interest. Considering a simple alkyl ketone in a protic solvent, for example, it can be seen that hydroxide ion and primary alkoxide ions will convert only a small fraction of such a ketone to its anion.
KINETIC FACTORS THERMODYNAMIC FACTORS 1.2. Regioselectivity and Stereoselectivity in Enolate Formation An unsymmetrical dialkyl ketone can form two regioisomeric enolates on deprotonation: In order to exploit fully the synthetic potential of enolate ions, control over the regioselectivity of their formation is required. Experimental conditions can often be chosen to provide a substantial preference for the desired regioisomer. So it is necessary to examine the process of enolate generation. may be governed by THE COMPOSITION OF AN ENOLATE MIXTURE
The enolate ratio is governed by kinetic control when the product composition is determined by the relative rates of the two or more competing proton-abstraction reactions.
The final composition of the mixture reflects the ratio of the (kinetic) rate constants, that is: UNSELECTIVE REACTION COMPARABLE RATES VERY DIFFERENT RATES SELECTIVITY
On the other hand, if enolates A and B can be interconverted readily, equilibrium is established and the product composition reflects the relative thermodynamic stability of the enolates. The enolate ratio is then governed by thermodynamic control.
The final composition of the mixture reflects the ratio of the (thermodinamic) stability constants, that is: COMPARABLE STABILITIES UNSELECTIVE REACTION VERY DIFFERENT STABILITIES SELECTIVITY
RAPID KINETIC CONTROL OF ENOLATE FORMATION QUANTITATIVE IRREVERSIBLE By adjusting the conditions under which an enolate mixture is formed from a ketone, it is possible to establish either kinetic or thermodynamic control. DEPROTONATION Ideal conditions for kinetic control of enolate formation are those in which deprotonation is rapid, quantitative and irreversible.
REGIOSELECTIVE GENERATION OF THE ENOLATE UNDER THERMODYNAMIC CONTROL REGIOSELECTIVE GENERATION OF THE ENOLATE UNDER KINETIC CONTROL EXPERIMENTAL CONDITIONS LESS STRONG BASE PROTIC SOLVENT STRONG BASE (LDA OR HMDS) LONG REACTION TIMES EXCESSKETONE APROTIC SOLVENT EXCESSBASE
Conditions of kinetic control usually favor the less substituted enolate. LESS SUBSTITUTED ENOLATE KINETIC CONTROL because the removal of the less hindered hydrogen is faster, for steric reasons, than removal of more hindered protons. Removal of the less hindered proton leads to the less substituted enolate. Steric factors in ketone deprotonation can be accentuated by using more highly hindered bases.
On the other hand, at equilibrium the more substituted enolate is usually the dominant species. MORE SUBSTITUTED ENOLATE THERMODYNAMIC CONTROL The stability of carbon-carbon double bonds increases with increasing substitution, and this effect leads to the greater stability of the more substituted enolate.
Same of the most studied chiral bases: It is also possible to achieve enantioselective enolate formation by using chiral bases. Enantioselective deprotonation requires discrimination between two enantiotopic hydrogens, such as in cis-2,6-dimethylcyclohexanone or 4-(t-butyl)cyclohexanone.
Enantioselective enolate formation can also be achieved by kinetic resolution,by preferential reaction of one of the enantiomers of a racemic chiral ketone such as 2-(t-butyl) cyclohexanone. Such enantioselective deprotonations depend upon kinetic selection bythe chiral base between prochiral or enantiomeric protons, that results in energy differences between diastereomeric transition states
Kinetically controlled deprotonation of a,b-unsaturated ketones usually occurs preferentially at the a' carbon adjacent to the carbonyl group. The polar effect of the carbonyl group is probably responsible for the faster deprotonation at this position. Under conditions of thermodynamic control, however, it is the enolate corresponding to deprotonation of the gcarbon that is present in the greater amount.
These isomeric enolates differ in stability in that the firstis fully conjugated, whereas the secondis cross-conjugated. In the cross conjugated enolate the delocalization of the negative charge is restricted to the oxygen and the a' carbon, whereas in the full conjugated enolate, the negative charge is delocalized on oxygen and both the a and the gcarbon.