Download

1 / 10
350 likes | 1.5k Views
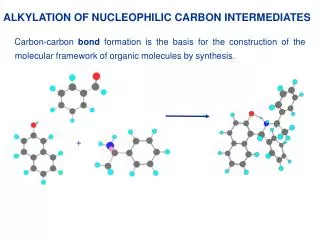
1.7. OXYGEN VERSUS CARBON AS THE SITE OF ALKYLATION. Enolate anions are ambident nucleophiles. Alkylation of an enolate can occur at either carbon or oxygen. Most of the negative charge of the enolate is on the oxygen atom, so it might be supposed that O-alkylation would dominate.

E N D
1.7. OXYGEN VERSUS CARBON AS THE SITE OF ALKYLATION Enolate anions are ambident nucleophiles. Alkylation of an enolate can occur at either carbon or oxygen. Most of the negative charge of the enolate is on the oxygen atom, so it might be supposed that O-alkylation would dominate. But not only charge density affect the C/O-alkylation ratio, and it is normally possible to establish reaction conditions that favor alkylation on carbon.
O-Alkylation is most pronounced when the enolate is dissociated. • In the polar aprotic solvent HMPA, the major product (83%) is the O-alkylated one. • In THF, where ion clustering occurs, all of the product is C-alkylated. • In t-butanol, where the acetoacetate anion is hydrogen-bonded by solvent, again only C-alkylation is observed.
Leaving-group effects on the ratio of C- to O-alkylation can be correlated by reference to the "hard-soft-acid-base" (HSAB) rationale. Of the two nucleophilic sites in an enolate ion, oxygen is harder than carbon. HARDER SOFTER Consequently, ethyl iodide, with the very soft leaving group iodide, reacts preferentially with the softer carbon site, rather than the harder oxygen. Oxygen leaving groups, such as sulfonate and sulfate, are harder, and alkyl sulfonates and sulfates react preferentially at the hard oxygen site of the enolate. C=O + C-C > C=C + C-O
Higher C/O-alkylation ratios are observed with alkyl halides than with alkyl sulfonates and sulfates. The highest C/O-alkylation ratios are obtained with alkyl iodides.
highly exotermic step with low activation energy TS EARLY TRANSITION STATE CHARGE DISTRIBUTION R Energy P favored by proceeds best with NUCLEOPHILIC SUBSTITUTION OF SN2 TYPE or SOFT - SOFT favored by TS P Energy PARTIAL BOND FORMATION LATE TRANSITION STATE R HARD - HARD COMBINATION OFNUCLEOPHYLE AND ELECTROPHYLE highly endotermic step
Therefore, conditions that favor a dissociated, more reactive enolate favor O-alkylation too. • TO SUMMERIZE • The amount of O-alkylation is maximized by use of an alkyl sulfate or alkyl sulfonate in a polar aprotic solvent. • The amount of C-alkylation is maximized by use of an alkyl halide in a less polar or protic solvent. • The majority of synthetic operations involving ketone enolates are carried out in THF or DME using an alkyl bromide or alkyl iodide, and C-alkylation is favored.
Intramolecular alkylation of enolates leads to formation of cyclic products. In addition to the other factors that govern C/O-alkylation ratios, the element of stereoelectronic control comes into play in such cases.
GEOMETRY REQUIRED FOR INTRAMOLECULAR C-ALKYLATION OF ENOLATE GEOMETRY REQUIRED FOR INTRAMOLECULAR O-ALKYLATION OF ENOLATE In order for C-alkylation to occur, the p orbital at the a carbon must be aligned with the C-Br bond in the linear geometry associated with the SN2 transition state. When the ring to be closed is six-membered, this geometry is accessible, and cyclization to the cyclohexanone occurs. With five-membered rings, colinearity cannot be achieved easily. Cyclization at oxygen then occurs faster than does cyclopentanone formation. The transition state for O-alkylation involves an oxygen lone-pair orbital and is less strained than the transition state for C-alkylation.
In enolates formed by proton abstraction from a,b-unsaturated ketones, there are three potential sites for attack by electrophiles: the oxygen, the a carbon, and the g carbon. The kinetically preferred site for both protonation and alkylation is the a carbon. Protonation of the enolate provides a method for converting a,b-unsaturated ketones and esters to the less stable b,g-unsaturated isomers. Alkylation also takes place selectively at the a carbon. The selectivity for electrophilic attack at the a carbon presumably reflects a greater negative charge, as compared with the g carbon.
Phenoxide ions are a special case related to enolate anions, with a strong preference for O-alkylation because C-alkylation disrupts aromatic conjugation. Phenoxides undergo O-alkylation in solvents such as DMSO, DMF, ethers, and alcohols. In water and trifluoroethanol, however, extensive C-alkylation occurs. These latter solvents form particularly strong hydrogen bonds with the oxygen atom of the phenolate anion. This strong solvation decreases the reactivity at oxygen and favors C-alkylation.