Download

1 / 18
180 likes | 203 Views
‘Protein sequencing’: Determining protein sequences. Why the protein sequence is important It determines the shape and function of the protein Why determining the sequence of a protein is important Can be used to identify the protein Can be used to find proteins with similar sequences

E N D
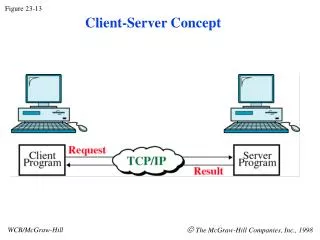
‘Protein sequencing’: Determining protein sequences • Why the protein sequence is important • It determines the shape and function of the protein • Why determining the sequence of a protein is important • Can be used to identify the protein • Can be used to find proteins with similar sequences • Useful for predicting the function of a new protein Figure 5-13
Approaches to protein sequencing • Chemical • Mass Spectrometry (tandem mass spec) • Genomic (DNA) data Figure 5-13
Key elements for chemical sequencing • Ability to fragment protein by various (sequence specific) enzymes or reagents • Separation of fragments by chromatography, for example • Ability to determine the amino acid sequences of fragments one amino acid at a time Figure 5-13
A approach for identifying the N-terminal amino acid: Dansyl chloride Note that if the sample contains more than one type of protein chain (e.g. connected together by disulfide bonds), then you will identify amino acids from both chains. Also note that a ‘blocked’ N-terminus (e.g. by acetylation) is a problem for chemical methods for sequencing. Also, hydrolysis (in the last step) degrades some amino acids (e.g. Trp, and converts amides to carboxylic acids). Text, Fig. 5.13 Figure 5-13
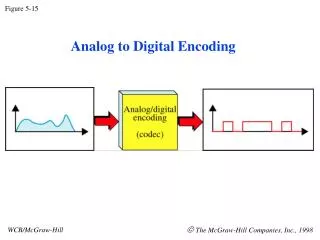
Edman degradation for (processive) sequencing, one amino acid at a time from the N-terminus Text, Fig. 5.15 Figure 5-15
Amino acid specificities for various cleavages used in sequencing Text, Table 5-3 Cyanogen bromide (chemical) Rn-1 = Met Table 5-3
Cleavage with multiple reagents gives overlapping fragments. This makes it possible to reconstruct the original sequence from sequences of the small fragments Text, Fig. 5.17 Figure 5-18
Cleavage with multiple reagents gives overlapping fragments. This makes it possible to reconstruct the original sequence from sequences of the small fragments Text, Fig. 5.12 Figure 5-18
Final stage: identifying any S-S bonds • Cleave protein into fragments (but without adding reducing reagent, so S-S bonds (if any) remain intact. • Separate fragments (e.g. chromatography). • (For each fragment) Cleave (i.e. reduce) disulfide bonds that might be present, then block the free thiols. • If a fragment had a disulfide bond, then sequence the fragment (or maybe just determine the amino acid composition). In favorable cases, this uniquely identifies which cysteines were disulfide bonded.
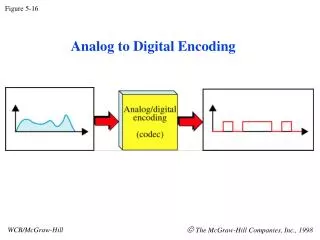
Mass spectrometry Moving charges are deflected from a straight line of flight by an applied magnetic field. The amount an ion is deflected, which is measured by the position where the molecular ion hits the detector, is a function of the sideways acceleration it experiences. F=ma, so the acceleration is a=F/m. The force is proportional to the charge, z. So, a z/m. So, mass spectrometry gives values for z/m (or m/z) for ionized molecules. Figure 5-16a
Example of a mass spectrum Note that the charge states are not measured as part of the experiment. They are shown here for explanatory purposes. Figure 5-16b
. . . . . These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Determining the mass of the parent ion from a mass spectrum: ‘textbook problem approach’ Choose a pair of adjacent peaks and let p=m/z for each peak. Then, Oops! Should be multiplication, not division. Figure 5-16b Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2,
. . . . . These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. These two linear equations can readily be solved for their unknowns, M and z. Solve the first equation for M. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Then plug this result into the second equation. Determining the mass of the parent ion from a mass spectrum: computational approach • Typical computational approach: • Consider every possible mass for the parent ion • For each possible parent mass, calculate the set of expected peak positions, m/z • Compare the list of expected peak positions to the observed spectrum to evaluate the fit a typical mass spectrum may have peaks that are weak and not sharply defined, so determining the values of p for two adjacent peaks may not be so easy. Figure 5-16b Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2, Plugging in the values for p1 and p2,
Tandem-mass spectrometry: a powerful tool for protein sequencing Mixture of peptide fragments (or whole proteins) Fragment the protein or peptide fragment into small (overlapping) pieces. Masses of small fragments can be used to determine the sequence of the component selected by first filter. Comparison to genomic (DNA) sequence data can be very important here.
Evolution of Protein Sequences: Aligned sequences of cytochrome c (from Dickerson) Text, Table 5-5
Evolution of Protein Sequences: Aligned sequences of cytochrome c (from Dickerson) From the first floor of Boyer Hall, courtesy of Dick Dickerson
Evolution of Protein Sequences: Comparison of protein sequences (1) reveals and (2) takes into account, substitution probabilities for amino acids. Note positive scores below for similar amino acids (i.e. conservative substitutions) Positive (good) score for glutamate (E) to aspartate (D) substitution Negative (bad) score for isoleucine (I) to arginine (R) substitution
Evolution of Protein Sequences: Comparison of protein sequences between different organisms has been extremely important in establishing evolutionary relationships between species Text, Fig. 5.21