Download

1 / 38
1.02k likes | 5.53k Views
ELECTROPHORESIS Definitions Theory of Electrophoresis Electrophoretic Technique General Procedures Types of Electrophoresis Technical Considerations Ref: Burtis & Ashwood; Tietz Fundamentals/Textbook. 1. DEFINITIONS Electrophoresis

E N D
ELECTROPHORESIS • Definitions • Theory of Electrophoresis • Electrophoretic Technique • General Procedures • Types of Electrophoresis • Technical Considerations Ref: Burtis & Ashwood; Tietz Fundamentals/Textbook
1. DEFINITIONS Electrophoresis • Migration of charged solutes in a liquid medium under an electrical field • Many biological molecules have ionisable groups eg. amino acids, proteins, nucleotides, nucleic acids • Under an electric field -> charged particles migrate to anode (+) or cathode (-)
Zone Electrophoresis • Migration of charged molecules • Support medium • porous eg. CA or agarose • can be dried & kept • Same pH & field strength thru’out • Separation based on electrophoretic mobility • Separates macromolecular colloids eg. proteins in serum, urine, CSF, erythrocytes; nucleic acids
Isotachophoresis • Migration of small ions • Discontinuous electrolyte system • leading electrolyte (L- ions) & • trailing electrolyte (T- ions) • Apply sample solution at interphase of L & T • Apply electric field -> each type of ion arrange between L and T ions -> discrete zones • Separates small anions, cations, organic & amino acids, peptides, nucleotides, nucleosides, proteins
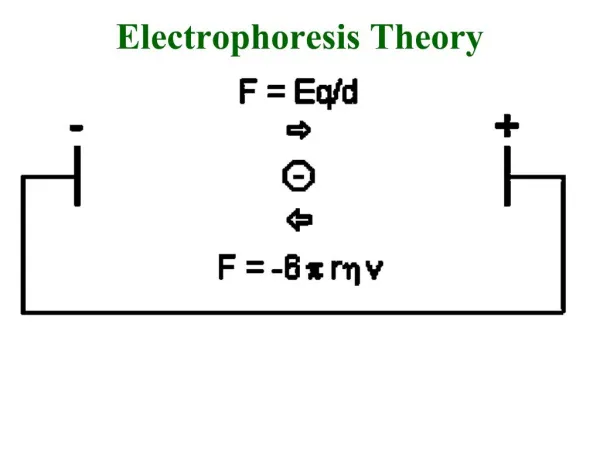
2. THEORY of ELECTROPHORESIS • Many biological molecules exist as (a) cations or (b) anions • Solution with pH < pI -> ampholyte/zwitterion has overall +ve charge • Solution with pH > pI -> ampholyte has overall –ve charge • Under an electric field -> cations/overall +ve migrate to cathode -> anions/overall -ve migrate to anode
Rate of migration depends on: • Net electrical charge of molecule • Size & shape of molecule • Electric field strength • Properties of supporting medium • Temperature of operation
3. ELECTROPHORETIC TECHNIQUE 3a. Instrumentation & Reagents • Buffer boxes with buffer plates -> holds buffer • Platinum or carbon electrode -> connected to power supply • Electrophoresis support -> with wicks to contact buffer • Cover -> minimize evaporation (Fig 7-1)
3b. Power Supplies • Power pack: supply current between electrodes • Flow of current -> Heat produced • increase in migration rate -> broadening of separated samples • formation of convection currents -> mixing of separated samples • thermal instability of heat sensitive samples • water loss -> concn of ions -> decrease of buffer viscosity -> decrease in resistance • To minimize problems: use constant-current power supply
3c. Buffers • To carry applied current & to fix the pH => determine electrical charge & extent of ionization => which electrode to migrate • Ionic strength of buffer • thickness of ionic cloud -> migration rate -> sharpness of electrophoretic zones • [ion] -> ionic cloud -> movement of molecules • Barbital buffers & Tris-boric acid-EDTA buffers
3d. Protein Stains • To visualize/locate separated protein fractions • Dyes: amount taken up depends on • Type of protein • Degree of denaturation of proteins by fixing agents Types of stains: Table 7-1
4. GENERAL PROCEDURES 4a. Separation • Place support material in EP chamber • Blot excess buffer from support material • Place support in contact with buffer in electrode chamber • Apply sample to support
cont. Separation • Separate component using constant voltage or constant current for length of time • Remove support, then -> dry or place in fixative -> treat with dye-fixative -> wash excess dye -> dry (agarose) or put in clearing agent (CA membs)
4b. Detection & Quantitation • Express as • % of each fraction present or • absolute concn • By densitometry • electrophoretic strip moved past an optical system • absorbance of each fraction displayed on recorder chart
5. TYPES OF ELECTROPHORESIS • Agarose Gel Electrophoresis b. Cellulose Acetate Electrophoresis c. Polyacrylamide Gel Electrophoresis d. Isoelectric Focusing e. Two-dimensional Electrophoresis
5a. Agarose Gel Electrophoresis (AGE) • Use agarose as medium • low concns -> large pore size • higher concns -> small pore size • Serum proteins, Hb variants, lactate dehydrogenase, CK isoenzymes, LP fractions • Pure agarose - does not have ionizable groups -> no endosmosis
Cont. AGE • Advantages: • low affinity for proteins • shows clear fractions after drying • low melting temp -> reliquify at 65oC • Disadvantage: • poor elasticity -> not for gel rod system -> horizontal slab gels
5b. Cellulose Acetate Electrophoresis (CAE) • Cellulose + acetic anhydride -> CA • Has 80% air space -> fill with liquid when soaked in buffer • Can be made transparent for densitometry • Advantages: • speed of separation • able to store transparent membranes • Disadvantages: • presoaking before use • clearing for densitometry
cont. CAE • Method: • wet CA in EP buffer • load sample about 1/3 way along strip • stretch CA in strips across a bridge • place bridge in EP chamber -> strips dip directly into buffer • after EP, stain, destain, visualise proteins • For diagnosis of diseases • change in serum protein profile
5c. Polyacrylamide Gel Electrophoresis (PAGE) • Tubular-shaped EP cell -> pour small-pore separation gel -> large-pore spacer gel cast on top -> large-pore monomer solution + ~3ul sample on top of spacer gel • Electrophoresis -> all protein ions migrate thru large-pore gels -> concentrate on separation gel -> separation due to retardation of some proteins
Average pore size in 7.7% PAGE separation gel about 5nm -> allow most serum proteins to migrate -> impedes migration of large proteins eg fibrinogen, 1-lipoprotein, 2-macroglobulin • Advantages: • thermostable, transparent, strong, chemically inert • wide range of pore sizes • uncharged -> no endosmosis • Disadvantages: • carcinogenic
Denaturing PAGE/SDS-PAGE • Boil sample for 5 mins in sample buffer containing -mercaptoethanol & SDS • -mercaptoethanol: reduce disulfide bridges • SDS: binds strongly to & denatures proteins • Proteins denatured -> opens into rod-shaped structures -> separate based on size • Use: • To assess purity of protein • To determine MW of protein
(ii) Native PAGE • Use non-denaturing conditions -> no SDS or -mercaptoethanol -> proteins not denatured • Proteins separate based on: • different electrophoretic mobilities • sieving effects of gel • Use • to obtain native protein/enzyme • to study biological activity
5d. Isoelectric Focusing • To separate amphoteric cpds eg. proteins • Proteins moves to zone where: pH medium = pI protein => charge = 0 • pI of protein confined in narrow pH range -> sharp protein zones • Method: • use horizontal gels on glass/plastic sheets • introduce ampholytes into gel -> create pH gradient
cont. IEF Method • apply a potential difference across gel • anode -> area with lowest pH • cathode -> area with highest pH • proteins migrate until it arrives at pH = pI • wash with fixing solution to remove ampholytes • stain, destain, visualise • Separations of proteins with 0.01 to 0.02pH unit differences (Fig 7-4)
5e. Two-Dimensional (2D) EP (ISO-DALT) • 1st D using IEF EP -> in large-pore medium -> ampholytes to yield pH gradient • 2nd D using molecular weight-dependent EP -> in polyacrylamide -> linear or gradient • O’Farrell method: • use -mercaptoethanol (1st D) & SDS (2nd D) • Detect proteins using Coomassie dyes, silver stain, radiography, fluorography • Separates 1100 spots (autoradiography)
6. TECHNICAL CONSIDERATIONS Electroendosmosis/Endosmosis • Support in contact with water -> adsorb hydroxyl ions -> negative charge • Negative charge on support attract positive ions in solution -> Stern potential • As distance from -ve charge surface -> no. of –ve ions -> zeta () potential -> eventually no. +ve ions = -ve ions (Fig 7-8)
When apply current to system -> -ve charges on support remain fixed -> cloud of ions in solution move to electrodes -> ions highly hydrated => as ionic cloud moves, solvent also moves • Movement of solvent relative to fixed support => endosmosis • Movement of water in one direction • Macromolecules moving in opposite direction oppose flow of hydrated +ve ions -> may remain immobile or be swept to opposite pole
cont. Endosmosis • In cellulose acetate & agarose gel • Reduce endosmosis by • removing/modifying charged groups on support • adding of sucrose or sorbitol -> increase osmolality
IMMUNOASSAYS • Basic Concepts & Definitions • Measurement of Antibody Affinity • Quantitative Methods – competitive & noncompetitive assays Ref: Burtis & Ashwood; Tietz Fundamentals/Textbook Jan Klein & Vaclav Horejsi; Immunology (1997) Coleman Lombard Sicard; Fundamental Imm (1992) Gary D. Christian; Analytical Chem (1994)
1. BASIC CONCEPTS & DEFINITIONS Immunoassay: use of antibodies to detect analyte 1a. Antibodies • Immunoglobulins that bind to Antigens • 5 classes: IgG, IgA, IgM, IgD, IgE 1b. Immunogen • Protein or a substance coupled to a carrier • When introduced into foreign host -> induce Ab to form 1c. Antigen • Any material which can react with Ab • May not induce Ab formation
1d. Antigen-Antibody Binding • Ab molecules have specific binding sites -> bind tightly to Ag -> cause pptn/neutralization/ death • Binding of Ag to Ab due to • van der Waals forces • hydrophobic interactions • charged group attractions • Can measure Antibody affinity: strength of binding between Ab & Ag
2. MEASUREMENT OF ANTIBODY AFFINITY • Binding of Ag to Ab is reversible -> association & dissociation Ag + Ab <-> AgAb • Law of mass action: Rate of rxn to concn of reactants ka[Ag][Ab] = kd[AgAb] K = ka/kd = [AgAb]/ [Ag][Ab] where K is equilibrium constant or affinity constant
r/c = nK – rK r = no. of molecules of bound Ag per Ab molecule c = concn of free Ag n = valency of Ab • Plot r/c vs r => Scatchard Plot • Straight line with slope k • x intercept gives n • y intercept gives nK • K (liters/mole) measures affinity of complex
Why measure Affinity of an Antibody? • To assess Ab specificity • It influences the functional efficiencies of Abs eg. high-affinity Abs are very dependable for various applications: • Diagnostic • Therapeutic • Analytical
3. QUANTITATIVE METHODS • Read & Understand from Tietz Fundamentals: • Radial Immunidiffusion Immunoassay • Electroimmunoassay • Turbidimetric & Nephelometric Assays • Labeled Immunochemical Assays
COMPETITIVE vs NONCOMPETITIVE RXNS A. Competitive Immunoassays • Used when have limited reagents (Ag) (i) Simultaneous Competitive Assay • Labels Ag (Ag*) & unlabeled Ag compete for binding to Ab • The probability of Ab binding to Ag* is inversely to [Ag] Ab + Ag + Ag* <-> Ab:Ag + A-Ag*
(ii) Sequential Competitive Assay • Step 1: unlabeled Ag mixed with excess Ab -> binding allowed to reach equilibrium • Step 2: Ag* added sequentially -> equilibrate • After separation -> det bound Ag* -> calculate [Ag] • Larger fraction of Ag bound to Ab than in simultaneous assay • If k1>> k2 -> in Ab:Ag -> in Ag* binding • Provide two- to four- fold improvement in detection limit
b. Noncompetitive Immunoassays • Used when have excess reagent • Immobilization of Ab to support • Passively adsorption or bind covalently • Direct or indirect attachment (Table 9-3) ii. Ag allowed to react with Ab -> wash other proteins iii. Add labeled Ab (conjugate) -> reacts with bound Ag • Determine bound label -> [Ag*] or its activity is [Ag]