Download

1 / 29
330 likes | 1k Views
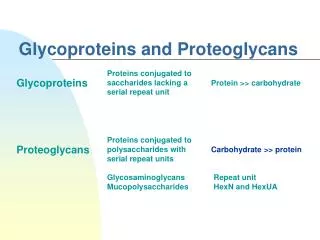
Glycosaminoglycans and Proteoglycans Dr. M. Jawad Hassan. Extracellular Macromolecules. macromolecule % carb . glycosaminoglycans * (GAGs) 100 proteoglycans * 90-95 glycoproteins 2-30 fibrous proteins 1-2 Examples of functions: mechanical support lubrication

E N D
Glycosaminoglycans and Proteoglycans Dr. M. Jawad Hassan
Extracellular Macromolecules macromolecule% carb. glycosaminoglycans* (GAGs) 100 proteoglycans* 90-95 glycoproteins 2-30 fibrous proteins 1-2 Examples of functions: mechanical support lubrication cushioning adhesives cell spacers selective filters *mucopolysaccharides, mucoproteins, respectively 1
GAG structure A sugar • exist as: • independent moleculese.g., hyaluronate & heparin • parts of larger structurese.g., in proteoglycans • heteropolysaccharides repeating structure: disaccharide (AB)nABABAB… • where A is usually 1 uronic acid (hexose with C6 as COO– ) • & B is 1 glycosamine (amino sugar) derivative • unbranched • glycosidic linkage • anomeric C of 1 unit linked to hydroxyl of adjacent unit B sugar 3
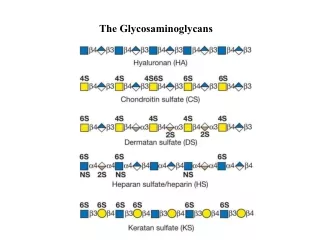
GAG structure: repeating units 4 GAG A sugar B sugarhyaluronate glucuronate N-acetyl glucosamine * 2 5
GAG structure: repeating units 4 GAG A sugar B sugarhyaluronate glucuronate N-acetyl glucosamine chondroitin sulfate glucuronate N-Ac galactosamine 4-SO4 dermatan sulfate iduronate " heparan sulfate glucuronate glucosamine N-SO3, 6-SO4 heparin iduronate 2-SO4 " keratan sulfate galactose N-Ac glucosamine 6-SO4 *opposite configuration in iduronate glucuronate/iduronate: epimersat C5 glucose/galactose: epimers at C4 * 2 5
Hyaluronate 5 • mol wt: 106 – 107 (5000 – 50,000 monosaccharide units) • very polar: 2 hydroxyls/unit 6 heteroatoms/unit COO– every other unit binds cations: Na+, Ca++ Display of HA in motion A B A B A B hyastk2.gif – – – 1 2 3 4 5 6 (glucuronate–N-acetyl glucosamine)3 (glcUA–glcNAc)3
Hyaluronate: structure & properties 6 • extended structure (charge repulsion) • hydrophilic: binds 10–100 × wt in H2O • additional, loosely associated H2O, so that volume occupied ~1000 × weight Display of HA with glcUAs in CPK hyacpk2.gif – – – 2 3 1 4 6 5 (glcUA–glcNAc)3 glcUAs in space-filling form (CPK)
Hyaluronate Alberts et al. Fig. 19-37 • solutions viscous, gel–like, compression-resistant • occurrence: EC matrix,esp. in developing tissue healing wounds synovial fluid • functions: lubricant shock absorber flexible cement attachment site path for cell migration • made by fibroblasts • degraded by hyaluronidase hyaluronidase • bacterial hyaluronidase facilitates spread of infection 7
Heparin • mol wt ~ 104 • ~ 40 monosaccharide units • made & released from mast cells in lungs & liver heparin cell 8
Heparin • mol wt ~ 104 • ~ 40 monosaccharide units • made & released from mast cells in lungs & liver • also associated with luminal surface of endothelium • anticoagulant • forms complex with antithrombin III • this complex binds to thrombin, inactivating it • as a result, clot growth is limited • fast-acting, making it therapeutically useful heparin cell 8
Extracellular Macromolecules macromolecule% carb. glycosaminoglycans* (GAGs) 100 proteoglycans* 90-95 glycoproteins 2-30 fibrous proteins 1-2 Examples of functions: mechanical support lubrication cushioning adhesives cell spacers selective filters * mucopolysaccharides, mucoproteins, respectively
Proteoglycans (PGs) • composed of as many as 200 GAG chains covalently bonded to a core protein via serine side chains • molecular weight range: 105 – 107 • GAG chains: chondroitin sulfate, heparan sulfate, dermatan sulfate, keratan sulfate Examples • decorin • many connective tissues • binds type I collagen, TGF-b • perlecan • basal laminae • structural & filtering function • aggrecan • syndecan (slide 13) from Alberts et al. Fig. 19-57 AlbertsT 19-3:Dcrn GAGchndSO4/drmSO4 GAG chains core protein 9
PG in basal lamina of renal glomerulus adapted from Alberts et al., 3 ed., Fig. 19-56 • network offibrousproteins &perlecanPG forms filter entactin perlecan GAG:heparan SO4 laminin type IV collagen 10
Proteoglycans: aggrecan • ~100 GAG chains/molecule • ~100 monosaccharides/GAG chain • each "bristle" = 1 GAG chain • each GAG chain is either chondroitin sulfate or keratan sulfate • GAG chains linked to ser side chains of core protein based onAlberts et al. Fig. 19-37 4ed. 19-40 core protein GAG chains 11
An aggregate of aggrecans & hyaluronan 1m • major GAG–PGin cartilage • link proteins bind non-covalently • with bound H2O,disperses shocks,compressive force • ~ cell size • adhesion proteins link to collagen & cells • degraded by chondroitin sulfatase, etc core protein link proteins hyalur-onan keratansulfate chondroitinsulfate 12 Alberts et al. Fig. 19-41
Repeating units of some common glycosaminoglycans of extracellular matrix linear polymers composed of repeating disaccharide units Glucoronic acid N-Acetylglucosamine
Proteoglycans: syndecan • cell-surface PG • core protein domains • intracellular • transmembrane • extracellular 5 GAGs attached • functions • interactions • cell-cell • cell-matrix • growth factor receptor GAG chains outside inside core protein Lehninger et al.Fig. 9-22 13
Proteoglycans are glycosaminoglycans-containing macromolecules of the cell surface and extracellular matrix Proteoglycan structure
GAG synthesis & breakdown –UDP • synthesis • activated precursors: UDP–monosaccharide derivativese.g., UDP–glucuronate • residues added one at a time in Golgi complex • sulfate moieties • degradation • lysosomes • specific glycosidases & sulfatases • mucopolysaccharidoses • genetic disorders • accumulation of GAG due to absence of a specific glycosidase or sulfatase – adenine – – – 14
GAG synthesis & breakdown • Synthesis of amino sugars • GlcNAc and GalNac (fructose 6 phosphate) • NANA (N-acetylemannosamin and phophoenolpyruvate) • CMP-NANA synthetase for activation.
Synthesis of acidic sugars • Glucoronic acid and L-Iduronic acid • Diet, lysosomal degradation via uronic acid pathway • Active form is UDP-glucoronic acid Synthesis of carbohydrate chain and addition of sulphates Xylosyltransferase sulphotransferases PAPS is sulphar donor
Degradation • Acid hydrolases • Phagocytosis • Lysosomal degradation (endoglycosidases)
Properties of proteoglycans • Glycosylated proteins which have covalently attached highly anionic glycosaminoglycans (GAGs) • Highly hydrated gels (due to charged sugars). Resist compression -Sulfated glycosaminoglycans (disaccharides) are negatively charged: bind cations and water • Core proteins link to hyaluronicacid (MW: 3 x 106) • Number of disaccharides typically found in each glycosaminoglycan chain Heparin/Heparan sulfate (n = 15-30) Keratan sulfate (n = 20-40) Chondroitin sulfate (n < 250) Hyaluron (n < 50,000) • Can be in the ECM and on the surface of cells E.g. Syndecan (integral membrane protein with Heparan sulfate) is present on the surface of epithelial cells
MUCOPOLYSACCHARIDOSES (MPS) Rare inborn errors in the degradation of glycosaminoglycans result in a series of diseases called mucopolysaccharidoses. They are characterized by mental retardation and/or structural defects. MPS Type I Hurler’s syndrome results from a deficiency of alpha-L-iduronidase. Heparan sulfate and dermatan sulfate accumulate. There is growth and mental retardation with characteristic facial changes. MPS Type II Hunters syndrome is similar to Hurler’s syndrome but the enzyme deficiency is for iduronate sulfatase and the inheritance is X-linked. MPS Type III Sanfilipo’s syndrome is caused by a deficiency of one of four enzymes of which three are hydrolases and one is an N-acetyltransferase. There is severe mental retardation but only mild structural features. Other MPS Types are IV, VI and VII. There is no MPS Type V.
MPS I (Hurler Syndrome) A deficiency of L-iduronidase leads to mental retardation and structural changes due to accumulation of dermatan sulfate and heparan sulfate
MPS II (Hunter Syndrome) X-linked disease due to a deficiency of iduronate sulfatase
MPS III (Sanfilippo Syndrome) Deficiency in one of four degradative enzymes leads to severe mental retardation but little structural change
MPS IV (Morquio Syndrome) Deficiency of a galactose-6-sulfatase or a beta-galactosidase leads to accumulation of keratan sulfate with normal intelligence but severe deformity