Download

1 / 25
270 likes | 633 Views
AMINO ACID METABOLISMS. Amino acid structure. Essential amimo acids Val, Leu Ile Phe Met, Thr, Lys, Arg * , Hys * , Trp Non essential amino acids Gly, Ala, Ser, Pro, Hyp 1 , Cys, Tyr, Asn, Gln, Asp, Glu, Hyl 1. AMINO ACID METABOLISM. BODY PROTEINS. Degradation. Proteosynthesis.

E N D
Essential amimo acids Val, Leu Ile Phe Met, Thr, Lys, Arg*, Hys*, Trp Non essential amino acids Gly, Ala, Ser, Pro, Hyp1, Cys, Tyr, Asn, Gln, Asp, Glu, Hyl1
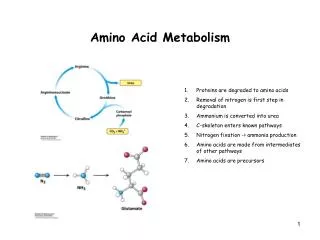
AMINO ACID METABOLISM BODY PROTEINS Degradation Proteosynthesis NONPROTEIN DERIVATIVES Porphyrins Purines Pyrimidines Neurotransmitters Hormones Komplex lipids Aminosugars Digestion AMINO ACIDS DIETARY PROTEINS GLYCOLYSIS KREBS CYCLE Conversion (Carbon skeleton) Transamination UREA NH3 GLUCOSE CO2 KETONBODIES
Enzymes cleaving the peptide bonds Endopeptidases – hydrolyse the peptide bond inside a chain Pepsin, trypsin, chymotrypsin Exopeptidases – split the peptide bond at the end of a protein molecule Aminopeptidase, carboxypeptidases Dipeptidases Pepsin (pH 1.5 – 2.5) – peptide bond derived fromTyr, Phe, bonds between Leu and Glu Trypsin (pH 7.5 – 8.5) – bonds between Lys a Arg Chymotrypsin (pH 7.5 – 8.5) – bonds between Phe a Tyr
General reactions of amino acids deamination a-keto acid NH2transaminationa-keto acid + amino acid R C H COOHdecarboxylation amine General reactions of amino acids are transamination and deamination of a-amino group Decarboxylation reaction gives biologically active amines
Transamination reactions The glutamate which is produced by these transaminase reactions is oxidatively deaminated by glutamate dehydrogenase to release ammonium:
Transamination reactions (enzymes aminotransferases) Transaminases are enzymeswhich transfer the amino groupfrom an amino acid to a keto acid, usually alpha-ketoglutarate, essentially swapping an amino group with a keto group: (Alanine-a-ketoglutarate transferase) another similar reaction yields more common products: (aspartate-a-ketoglutarate transferase)
These reactions are mediated by pyridoxal phosphate (PLP), a derivative of pyridoxine (vitamin B6):
pyridoxal phosphate pyridoxamine phosphate pyridoxamine phosphate pyridoxal phosphate
Clinicaly important transaminases ALT Alanine-a-ketoglutarate transferase Clinical marker for irreversibile liver damage AST aspartate-a-ketoglutarate transferase Clinical marker for irreversibile myocardial damage
Amino acids with three carbons are converted to pyruvate: • Alanine • Serine • Cysteine Serine is deaminated by serine dehydratase to form pyruvate + NH4+ in a reaction which doesn't involve the transaminase but does use pyridoxal phosphate (PLP) as a reactive group. Similarly, threonine can be dehydrated and deaminated to yield pyruvate. Glycine can be converted to serine for degradation, or it can be cleaved to release CO2, NH4+ and an activated one-carbon unit. In addition, three carbons from tryptophan are converted to pyruvate by way of alanine.
Amino acids with five carbons are converted to alpha-ketoglutarate: • Arginine • Glutamine • Histidine • Proline These amino acids are first converted to glutamate which is transaminated to alpha-ketoglutarate.
Some amino acids are converted to succinyl-CoA: • Methionine • Valine • Isoleucine These amino acids are converted to propionyl-CoA which is carboxylated to methylmalonyl-CoA which is converted to succinyl-CoA. This last step is an isomerization catalyzed by Methylmalonyl-CoA mutase, an enzyme which uses cobalamin (vitamin B12).
Leucine and Lysine are converted to acetyl-CoA and acetoacetate: Degradation of valine, leucine, and isoleucine requires the oxidative decarboxylation of an alpha-keto acid. If this enzyme is defective, these acids accumulate in the blood and urine, resulting in maple syrup urine disease (branched chain ketoaciduria). This disease is characterized by physical and mental retardation.
Phenylalanine and Tyrosine are converted to fumarate and acetoacetate A serious disease results from the inability to oxidize phenylalanine by a defective phenylalanine hydroxylase. This results in high levels of phenylpyruvate developing (phenylpyruvate is the result of transamination of phenylalanine with an amino acid). The disease is phenylketonuria (PKU), and results in severe mental retardation and shortens the life span so that half the carriers are dead at 20 and 75% are dead at 30 if it is untreated. It is a genetic disorder and can result from aberrant splicing of the normal phenylalanine hydroxylase transcript. Therapy for the disease involves restricting the intake of phenylalanine, and must be started immediately after birth. Screening for the disease occurs at birth so that as many effects as possible can be avoided.
The twenty common amino acids are degrade to a total of seven different compounds, all of which are related to the citric acid cycle: Degradation of aminoacids gives intermediates for saccharides and lipides synthesis
Interconversion of amino acids and intermediates of carbohydrate metabolism
Amino acids Glucogenicketogenicglucogenic + ketogenic Ala Hyp Leu Ile Arg Met Lys Asp Pro Phe Cys Ser Trp Glu Thr Tyr Glu Val Gly His
Enzymes which metabolised amino acides containe vitamines as cofactors Vater soluble vitamins B THIAMINE B1 (thiamine diphosphate) oxidative decarboxylation of a-ketoacids RIBOFLAVIN B2 (flavin mononucleotide FMN, flavin adenine dinucleotide FAD) oxidses ofa-aminoacids NIACIN B3 – nicotinic acid (nikotinamide adenine dinucleotide NAD+ nikotinamide adenine dinukleotide phosphate NADP+) dehydrogenases, reductase PYRIDOXIN B6 (pyridoxalphosphate) transamination reaction and decarboxylation FOLIC ACID (tetrahydropholate) Meny enzymes of amino acid metabolism
Nitrogenous derivatives of amino acids Glycine heme, purine, creatine, conjugation of bile acids Histidine histamine Ornithine a arginin creatine, polyamines (spermidine, spermine) Tryptophan serotonine (melatonine) Tyrosine Epinephrine, norepinephrine Glutamic acid g-aminobutyric acid (GABA)
Aspartame (NutraSweet) consists of a methly ester of L-aspartate and L-phenylalanine: