Download
1 / 28
300 likes | 567 Views
HEMOSTASE. Fase vascular (vasoconstrição) Fase plaquetária (formação do trombo de plaquetas) Fase plasmática (formação do coágulo de fibrina). FASE PLAQUETÁRIA. Colagénio-f. von Willebrand (e f. I). Adesão. Gp membrana. Reacção de libertação. Colagénio, trombina, adrenalina, ADP

E N D
HEMOSTASE • Fase vascular(vasoconstrição) • Fase plaquetária(formação do trombo de plaquetas) • Fase plasmática(formação do coágulo de fibrina)
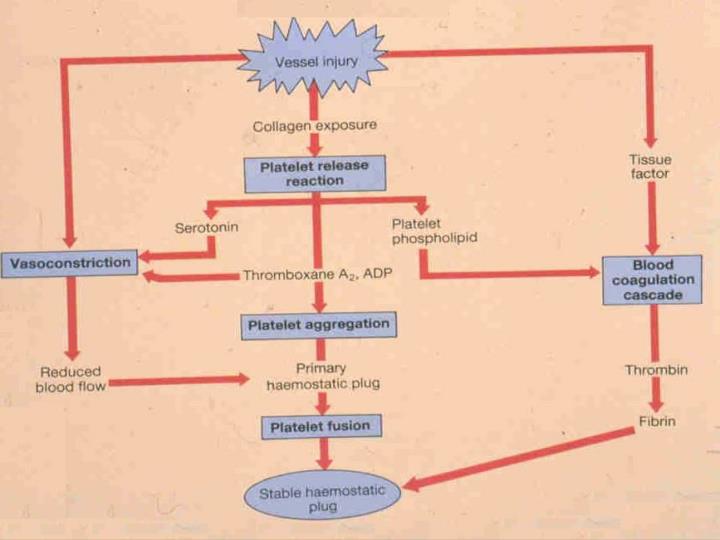
FASE PLAQUETÁRIA Colagénio-f. von Willebrand (e f. I) Adesão Gp membrana Reacção de libertação Colagénio, trombina, adrenalina, ADP (<AMP cíclico – trombostenina – secreção) primária ADP libertado Agregação agregação secundária (irreversível) Fusão Retracção do coágulo
FASE PLASMÁTICA • Via intrínseca(desencadeada pelo contacto com superfície estranha) • Via extrínseca(tromboplastina tecidular) • convergem as 2 vias navia comum • Fibrinólise
FASE PLASMÁTICA Via intrínseca (APTT) Via extrínseca (t. protrombina) Contacto com superfície estranha F. XIIa + pré-calicreína + quininogénio F. XII Tromboplastina tecidular F. XI F. XIa F. VII Ca++ Ca++ F. IX F. IXa + F. VIII + PF3 Ca++ F. X F. Xa F. X Via comum (t. trombina)
FASE PLASMÁTICA(via comum) F. Xa F. V + PF3 Ca++ Protrombina Trombina Fibrinogénio Fibrina Fibrina polimerizada (solúvel em ureia) F. XIII Fibrinólise Produtos de degradação da fibrina Fibrina insolúvel (cross-linked)
PERTURBAÇÕES DA HEMOSTASEANAMNESE e EXAME FÍSICO HISTÓRIA • Hemorragia ou púrpura ? Espontânea ou provocada ? • Hemorragia imediata e transitória ou retardada e prolongada ? • Hemorragia excessiva de pequenos cortes ? • Hematomas ou equimoses espontâneas grandes e frequentes ? • Pesquisa de doença sistémica subjacente • História familiar ? Padrão de transmissão • Ingestão de fármacos
PERTURBAÇÕES DA HEMOSTASEANAMNESE e EXAME FÍSICO EXAME FÍSICO • Manifestações purpúricas da pele (petéquias, equimoses) • Hemorragia das mucosas (deficiência das plaquetas ou fibrinólise, telangiectasia, factores locais) • Hemartrose e anquilose (deficìência de factores VIII ou IX) • Estigmas de doença hepática • Vasculite, conectivite (Ehlers-Danlos, “easy bruising”, púrpuras senil, dos corticoides, do escorbuto, de Henoch-Schonlein, associada a infecções)
MANIFESTAÇÕES PURPÚRICAS t. sangria > baixo nº plaquetas normal ou elevado Púrpura trombocitopénica Disfunção plaquetária est. agreg. plaq. M.O. Mk < adquirida congénita Mk > causa periférica causa central B-Soulier W-Aldrich Ch-Higashi Tr. Glanzman von Willebrand procurar Macroglobulinémia Dextrano Drogas Aplasia distrombopoiese esplenomegalia atcs antiplaquetários drogas, LE, HIV estudo da coagulação hemólise PTI, p. iatrogénica, LE, SIDA CID, SHU, PTT, congénitas
Fármacos com acção anti-agregante • Aspirina, dipiridamol, indometacina, sulfinpirazona • Fenilbutazona, AINE • Penicilinas, cefalosporinas • Fenotiazinas, anti-depressivos tricíclicos • Clofibrato, cloroquina • Prostaglandinas, etc
TROMBOCITOPENIA DE CAUSA IATROGÉNICA • De causa central - mielosupressores - hidroclorotiazida - etanol - estrogénios • De causa periférica - causalidade comprovada(sulfamidas, quinidina, frutos citrinos, feijão, carbamazepina, DFH, fenobarbital, meprobamato, paracetamol, fenilbutazona, cefalotina, PAS, digitoxina, metildopa, arsenicais) - causalidade suspeita(aspirina, cloropropamida, cloroquina, clorotiazidas, sais de ouro, insecticidas, etc)
PÚRPURA TROMBOCITOPÉNICA IDIOPÁTICA ou IMUNE • Forma aguda ou infantil • Forma crónica ou adulta TRATAMENTO • Repouso no leito • Vigilância de hemorragias • Corticosteroides • Ig i.v. • Esplenectomia • Antifibrinolíticos • Imunosupressores • Transfusão de plaquetas (?)
TRANSFUSÃO DE PLAQUETAS • Falência de produção - leucemias agudas - aplasia medular • Perda de plaquetas - transfusões maciças - CEC • Consumo - DIC • Trombocitopenias hereditárias(W-A, B-S) • Trombastenia e disfunção plaquetária(?) • Trombocitopenia natal isoimune





