Download
1 / 1
10 likes | 26 Views
Understanding network robustness is crucial for studying human diseases. This study presents a method using graph spectrum analysis to assess network changes with minimal node removal. Application on yeast protein interaction network shows correlation with functional modules, suggesting potential for genetic interaction screening and drug target identification. Results indicate the method's efficacy in simulating network robustness. Future directions involve predictive models based on graph spectral change, connectivity, and modularity. The Laplacian spectrum method uses network eigenvalues to measure topological changes accurately, making it a valuable tool for studying network robustness and potential drug target identification.

E N D
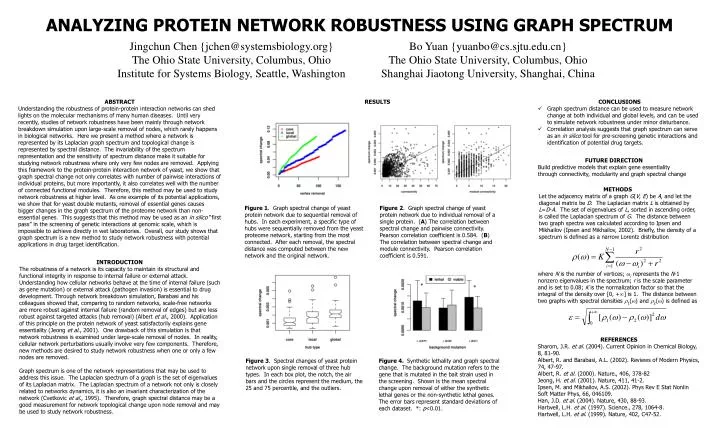
Jingchun Chen {jchen@systemsbiology.org} The Ohio State University, Columbus, Ohio Institute for Systems Biology, Seattle, Washington Bo Yuan {yuanbo@cs.sjtu.edu.cn} The Ohio State University, Columbus, Ohio Shanghai Jiaotong University, Shanghai, China ABSTRACT Understanding the robustness of protein-protein interaction networks can shed lights on the molecular mechanisms of many human diseases. Until very recently, studies of network robustness have been mainly through network breakdown simulation upon large-scale removal of nodes, which rarely happens in biological networks. Here we present a method where a network is represented by its Laplacian graph spectrum and topological change is represented by spectral distance. The invariability of the spectrum representation and the sensitivity of spectrum distance make it suitable for studying network robustness where only very few nodes are removed. Applying this framework to the protein-protein interaction network of yeast, we show that graph spectral change not only correlates with number of pairwise interactions of individual proteins, but more importantly, it also correlates well with the number of connected functional modules. Therefore, this method may be used to study network robustness at higher level. As one example of its potential applications, we show that for yeast double mutants, removal of essential genes causes bigger changes in the graph spectrum of the proteome network than non-essential genes. This suggests that this method may be used as an in silico “first pass” in the screening of genetic interactions at genomic scale, which is impossible to achieve directly in wet laboratories. Overall, our study shows that graph spectrum is a new method to study network robustness with potential applications in drug target identification. • CONCLUSIONS • Graph spectrum distance can be used to measure network change at both individual and global levels, and can be used to simulate network robustness under minor disturbance. • Correlation analysis suggests that graph spectrum can serve as an in silico tool for pre-screening genetic interactions and identification of potential drug targets. RESULTS FUTURE DIRECTION Build predictive models that explain gene essentiality through connectivity, modularity and graph spectral change METHODS Let the adjacency matrix of a graph G(V, E) be A, and let the diagonal matrix be D. The Laplacian matrix L is obtained by L=D-A. The set of eigenvalues of L, sorted in ascending order, is called the Laplacian spectrum of G. The distance between two graph spectra was calculated according to Ipsen and Mikhailov (Ipsen and Mikhailov, 2002). Briefly, the density of a spectrum is defined as a narrow Lorentz distribution Figure 1. Graph spectral change of yeast protein network due to sequential removal of hubs. In each experiment, a specific type of hubs were sequentially removed from the yeast proteome network, starting from the most connected. After each removal, the spectral distance was computed between the new network and the original network. Figure 2. Graph spectral change of yeast protein network due to individual removal of a single protein. (A) The correlation between spectral change and pairwise connectivity. Pearson correlation coefficient is 0.584. (B) The correlation between spectral change and module connectivity. Pearson correlation coefficient is 0.591. INTRODUCTION The robustness of a network is its capacity to maintain its structural and functional integrity in response to internal failure or external attack. Understanding how cellular networks behave at the time of internal failure (such as gene mutation) or external attack (pathogen invasion) is essential to drug development. Through network breakdown simulation, Barabasi and his colleagues showed that, comparing to random networks, scale-free networks are more robust against internal failure (random removal of edges) but are less robust against targeted attacks (hub removal) (Albert et al., 2000). Application of this principle on the protein network of yeast satisfactorily explains gene essentiality (Jeong et al., 2001). One drawback of this simulation is that network robustness is examined under large-scale removal of nodes. In reality, cellular network perturbations usually involve very few components. Therefore, new methods are desired to study network robustness when one or only a few nodes are removed. Graph spectrum is one of the network representations that may be used to address this issue. The Laplacian spectrum of a graph is the set of eigenvalues of its Laplacian matrix. The Laplacian spectrum of a network not only is closely related to networks dynamics, it is also an invariant characterization of the network (Cvetkovic et al., 1995). Therefore, graph spectral distance may be a good measurement for network topological change upon node removal and may be used to study network robustness. where N is the number of vertices; i represents the N-1 nonzero eigenvalues in the spectrum; r is the scale parameter and is set to 0.08; K is the normalization factor so that the integral of the density over [0, +] is 1. The distance between two graphs with spectral densities 1() and 2() is defined as * * REFERENCES Sharom, J.R. et al. (2004). Current Opinion in Chemical Biology, 8, 81-90. Albert, R. and Barabasi, A.L. (2002). Reviews of Modern Physics, 74, 47-97. Albert, R. et al. (2000). Nature., 406, 378-82 Jeong, H. et al. (2001). Nature, 411, 41-2. Ipsen, M. and Mikhailov, A.S. (2002). Phys Rev E Stat Nonlin Soft Matter Phys, 66, 046109. Han, J.D. et al. (2004). Nature, 430, 88-93. Hartwell, L.H. et al. (1997). Science., 278, 1064-8. Hartwell, L.H. et al. (1999). Nature, 402, C47-52. Figure 3. Spectral changes of yeast protein network upon single removal of three hub types. In each box plot, the notch, the air bars and the circles represent the medium, the 25 and 75 percentile, and the outliers. Figure 4. Synthetic lethality and graph spectral change. The background mutation refers to the gene that is mutated in the bait strain used in the screening. Shown is the mean spectral change upon removal of either the synthetic lethal genes or the non-synthetic lethal genes. The error bars represent standard deviations of each dataset. *: p<0.01. ANALYZING PROTEIN NETWORK ROBUSTNESS USING GRAPH SPECTRUM