Download

1 / 38
380 likes | 551 Views
Predictive rule inference for epistatic interaction detection in GWAS. Antonio Sze -To, 31 st January, 2013, Regular meeting. Genome-wide A ssociation Study (GWAS). DNA Sequencing. 3x 10 9 nucleotides. Case (with disease). DNA Sequencing.

E N D
Predictive rule inference for epistatic interaction detection in GWAS Antonio Sze-To, 31st January, 2013, Regular meeting
Genome-wide Association Study (GWAS) DNA Sequencing 3x 109 nucleotides Case (with disease) DNA Sequencing GWAS focus on associations between genetic variations (single-nucleotide polymorphisms) and traits like major diseases. Control (without disease) DNA Sequencing http://www.sodahead.com http://www.biol.unt.edu/~jajohnson/DNA_sequencing_process http://www.illustrationsof.com/63130-royalty-free-human-factor-clipart-illustration
Outline • Background • Motivation • Methodology • Experiment • Discussion • Conclusion
Background • Nucleotide: A, C, G, T • DNA: A sequence of nucleotides (e.g. ACGTTA) • Single Nucleotide Polymorphism (SNP) • Represents a kind of genetic variation DNA 1: …AACCGGTTAACCAGAAA… DNA 2: …AACCGGGTAACCCGAAA… DNA 3: …AACCGGGTAACCCGAAA… DNA 4: …AACCGGGTAACCCGAAA… Locus-X Allele: a variant of DNA sequence at a given position (locus). For simplicity, on a SNP, use capital letters, e.g. ‘A’, to represent major allele; use lowercase letters, e.g. ‘a’, to represent minor allele; SNP-1 Major Allele: G Minor Allele: T
Background • Human has 2 homozygous (the same) copies of each chromosome (a organized structure of DNA), usually one from the mother and one from the father. • If there are variants, here are the 3 cases: • Homozygous reference genotype (AA) • Heterozygous genotype (Aa) • Homozygous variant genotype (aa) Chromosome CSCI6200 Project Presentation
Definition (GWAS) Input: • Encoding • Homozygous reference genotype (AA) -> 0 • Heterozygous genotype (Aa) -> 1 • Homozygous variant genotype (aa) -> 2 Output: SNPs associated with diseases 1- order: {SNP1}, {SNP2}, {SNP3}… 2-order : {SNP1, SNP2}… 3-order : {SNP1, SNP2, SNP3}… …
Motivation • Complex diseases like cancer are likely to be associated with high-order SNP-SNP interactions but not single order. • For J SNPs, J2 pairs are needed to be considered for 2-way interactions. In general, for J SNPs and K-way interactions, there are O (JK) candidates for interactions. • It is computationally infeasible to examine all possible interactions. (J may be as large as 500, 000 in nowadays studies) • Here, we explore the use of smarter algorithms.
Concept Difficulty • Marginal effects are commonly used in practice to quantify the effect of individual variables on an outcome of interest. • E.g.: y = x1 + x2 + x3, what is the amount of change in Y that will be produced by a 1-unit change in x1? i.e. The marginal effect of x1. • Epistasis means that different genetic loci combine to cause disease, where marginal effects are not demonstrated. • From a data mining point of view, SNPs need to be considered jointly in learning algorithms rather than individually because the mapping between the attributes and class is nonlinear
Association Rule Mining - Identify associated patterns among items. E.g. {Butter, Bread} -> {Milk}
Predictive Rule Learning - Identify forward associations between SNPs and Class. E.g. {SNP1 = 1 ∩ SNP2 = 0 } -> 1
Definition • Definition 1 (Literal) • A sample S satisfies iff its i-th SNP has the value v. • Definition 2 (Predictive Rule) • A sample S satisfiesiffS satisfies all n literals and a class label . Example 1 S1 satisfies s=(2,2) S2 does not. Example 2 : s1 = (2,2) s2 = (3,3) -> 1 S1 satisfies S2 does not.
Definition • Definition 3 (Literal Relevance) • Given, • A literal in r is relevant iff • Definition 4 (Closed rule) • iff there is no literal such that
Definition • Problem Definition Given a dataset M = (), our goal is to find all closed predictive rules {()}, where U(()) >= T. T is a user-defined threshold to remove weak rules. We want to find out But not
Exhaustive search? How do we know the rule is closed? i.ethere is no literal such that If we have no prior knowledge, we may still need to test ALL possible combinations to prove the rule is closed. n literals and k SNPs
Exhaustive search? No ! • It is proved (by the authors) there is a upper bound
Utility Function U and its upper bound ,where R = , , Proof: , which is proportional to chi-square value
Utility Function U and its upper bound a = 200 c = 0 d = 200 - b is monotonically increasing with decreasing
Utility Function U and its upper bound The only way to achieve this is to add a new SNP with a specified genotype, which reduces 𝛿 Contingency Table for N(sj = gi) Contingency Table for N(r)
Utility Function U and its upper bound Contingency Table for N(sj = gi) Contingency Table for N(r)
Algorithm simulation (Conceptual) Given samples, class labels, maximum rule length L, a minimum threshold T, find out all predictive rules with U() >= T and number of literals <= L. Step 1: Compute contingency table T for each SNP Number of tables = 3 * number of SNPs Step 2: Add all literals into a list with checking
Algorithm simulation (Conceptual) Step 3: Grow the rule for each literal on the list Accept the growth only if Step 4: Stop when the depth of literals equals L Step 5: Generate the predictive rules and compute their corresponding chi-square values Given samples, class labels, maximum rule length L, a minimum threshold T, find out all predictive rules with U() >= T and number of literals <= L.
Experiments • Simulated data, compared with BEAM on statistical power • Models with marginal effects • Models without marginal effects • Large-scale simulation experiments • Real data, compared with BEAM and existing literatures • 3 WTCCC datasets • Bipolar disease [狂躁] • Crohn’s disease [克隆氏症] • Rheumatoid arthritis [類風濕性關節炎]
1. Models with marginal effects Source: Marchini, J., Donnelly, P., & Cardon, L. R. (2005). Genome-wide strategies for detecting multiple loci that influence complex diseases. Nature genetics, 37(4), 413-417.
1. Models with marginal effects SNPRuler (R) outperforms BEAM (B) in 4 models except one case.
2. Models without marginal effects Models with Non-linear mapping among SNPs (Needle-in-a-haystack) Examples of models withoutmarginal effects: SNP1 = Aa XOR SNP2 = Bb Non-linear mapping Examples of models with marginal effects: Linear mapping Source: Moore, J., & White, B. (2007). Genome-wide genetic analysis using genetic programming: The critical need for expert knowledge. Genetic Programming Theory and Practice IV, 11-28.
2. Models without marginal effects SNPRuler (R) outperforms BEAM (B) in comparisons.
3. Large-scale simulation experiments Ten times before
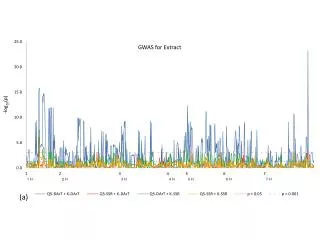
Results on WTCCC data • Both SNPRuler and BEAM are tested on these three WTCCC datasets. (∼500K SNPs, ~2000-~2000 study) • Bipolar disease [狂躁] • Crohn’s disease [克隆氏症] • Rheumatoid arthritis [類風濕性關節炎] • Comparison (CPU: Intel 3.0 GHz and RAM 8 GB)
Results on WTCCC data Rheumatoid Arthritis SNP Groups: (rs2809345, rs2808250) Individual P-values: (0.012, 0.299) Interaction P-value: 4.772 x 10-26 P-values have not been corrected for multiple hypothesis testing.
Results on WTCCC data Bipolar disease SNP Groups: (rs4844637, rs4844639) Individual P-values: (0.001, 0.774) Interaction P-value: 1.110x 10-16 P-values have not been corrected for multiple hypothesis testing
Conclusion • GWAS is motivated by its application to identify disease-associated biomarkers. • It is challenging because of the tremendous amount of possible SNP interactions and their non-linear mapping • Rule learning is a promising approach to quickly identify SNP-SNP interactions in a genome-wide scale. • One possible extension is to learn disjunctive predictive rules. (e.g. SNP1 = 1 OR SNP2 = 2).
Laboratory for Bioinformatics and Computational Biology, HKUST
Laboratory for Bioinformatics and Computational Biology, HKUST
Thank you ! Q & A