Download

1 / 6
70 likes | 674 Views
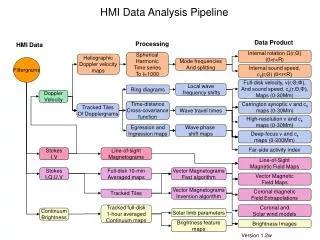
GWAS Analysis Pipeline. Less time Less errors Easier. Module 1 Module 2 Module 3 …. GWAS Data. Analysis Options (universal interface). Results. Input (data + analysis options). job2. job1. …. Job N. Pipeline Structure. gwaOption_R => R module. gwaOption_sas => SAS module.

E N D
GWAS Analysis Pipeline • Less time • Less errors • Easier Module 1 Module 2 Module 3 … GWAS Data Analysis Options (universal interface) Results
Input (data + analysis options) job2 job1 ….. Job N Pipeline Structure gwaOption_R => R module gwaOption_sas => SAS module gwaOption_C => C module Perl, Java, Matlab, … Result 1 Result 2 ….. Result N Final Results
gwas.shoptions.gwa gwas.R job2 job1 ….. Job N An Example sas -sysparm ‘optionfile.jobi’glm.sas (single machine mode) bsub –J jobnamesas -sysparm ‘optionfile.jobi’glm.sas (cluster mode) Result 1 Result 2 ….. Result N Final Results
[DATA] database=SAS genotype_dir=/dsg1/gwas/fhsgeno genotype_file=chr6_2 phenotype_dir=/dsg1/gwas/fhspheno phenotype_file=fhs100 markerinfo_dir= /dsg1/gwas/fhsmap markerinfo_file=mapall marker_selection=MAF<0.01 pedigree_dir= /dsg1/gwas/fhspedi pedigree_file=pediall subjectID=subject pedgreeID=famid markername=snp globbing=*/1:22 or 1,2,3,5:10 [ANALYSIS] phenolist_file= pheno_list=bmi/qt covariates= program=SASGLM analysis=mixed [OUTPUT] output_dir=/dsguser/qunyuan/fhs/bmi output_file= output_replace=no [RUN] clusterjobname=bmimixed memsize=1000M … #!/bin/sh OPFILE=$1 R --no-save < /dsguser/qunyuan/gwas/code/gwas.R $OPFILE > gwas.rlog & genotype_dir=IN_FILE genotype_file=genolist.txt gwas.sh options.gwa Pheno type covar program analysis run Bmi qt age,sex SASGLM mixed YES Obes ql NA SASGLM gee YES HD ql age SASGLM gee NO Age … Sex … … Program language location Maintainer SASGLM SAS /dsg1/code/sas/glm.sas Q.Zhang GSTAT R /dsg1/code/R/gstat.RQ.Zhang sasPLINK SAS /dsg1/code/sas/plink.sas J. Czajkowski …
%let genotype_dir=/dsguser/qunyuan/gwas/cluster/impudata/; %let genotype_file=mldosechr6_12; %let phenotype_dir=/dsguser/qunyuan/gwas/cluster/data/; %let phenotype_file=pheno100; %let markerinfo_dir=; %let markerinfo_file=; %let marker_selection=MAF<0.01; %let pedigree_dir=; %let pedigree_file=; %let subjectID=subject; %let pedgreeID=famid; %let markername=snp; %let phenolist_file=; %let phenolist=bmi; %let phenodata_type=qt; %let covariates=; %let program=SASGLM; %let analysis=mixed; %let output_dir=/dsguser/qunyuan/gwas/run/run2/mixck/; %let output_replace=yes; %let clusterjobname=; %let memsize=1000M; source("/dsguser/qunyuan/gwas/code/gwaslib.R") option.file=commandArgs()[3] read.table(option.file,sep="=",stringsAsFactors=F)->x x$V1=gsub(" ","",x$V1) x$V2=gsub(" ","",x$V2) rownames(x)=x$V1 gf=x["genotype_file",2] if (nchar(gf)>0) strsplit(gf,split=",")[[1]]->gfs if (nchar(gf)==0) { gfs=dir(x["genotype_dir",2]) if (x["output_replace",2]=="no") { outfs=dir(x["output_dir",2]) gfs=gfs[!(gfs %in% outfs)] } gfs=gsub(".sas7bdat","",gfs) } (dir.create(x["output_dir",2],recursive=T)) for (fi in gfs) { x["genotype_file",2]=fi xi=paste("%let ", x$V1,"=", x$V2, ";",sep="") optfile=paste(fi,".opt",sep="") write.table(xi,file=optfile,quote=F,row.names=F,col.names=F) cmd=paste("sas -memsize",x["memsize",2],"-sysparm",paste("'",optfile,"'",sep=""),x["program",2]) if (nchar(x["clusterjobname",2])>0) cmd=paste ("bsub -J",x["clusterjobname",2],cmd) system(cmd) } sasOption.job1 sasOption.job2 sasOption.job3 … … sasOption.jobN gwas.R sas –memsize 1000 M-sysparm ‘sasOption.job1’ /dsg1/code/sas/glm.sas & bsub –J jobname sas –sysparm ‘sasOption.job1’ /dsg1/code/sas/glm.sas %include “&sysparm”; . . .
Programming Tips Get all information that your program needs from one option file, pass the option file to your program via command line (e.g. sas –sysparm ‘optionfile’ yourscript.sas), or at least define all the options using variables at the top your script. For any information that may change in different analyses, instead of fixed names, use variables. (e.g. set my data(keep=&markname) ) Keep a good structure, use functionally independent macros and/or functions to organize your code. Don’t put every thing in one macro/function. %MACRO main(); %macro1(); *get p value for markers; %macro2(); *merge with marker positions & allele frequencies; %MEND main; %main(); Avoid name conflict. Name your variables and datasets using unique prefixes (e.g. QZ_temp). Write as little as you can. chrom loop; pheno loop; geno loop; yourmacro(indat=,y=,x=,covar=,method=,outdat=); end;end;end; Error handling.