Download
1 / 2
20 likes | 79 Views
Investigating surface energy difference vs. strain stability and evolution of nanostructures through VASP simulations at Brown University MRSEC. Exploring Si/Ge(001) surfaces in the context of quantum dots self-assembly. New insights into surface reconstruction, step formation, and strain. Integration of results into morphological evolution analysis methods.

E N D
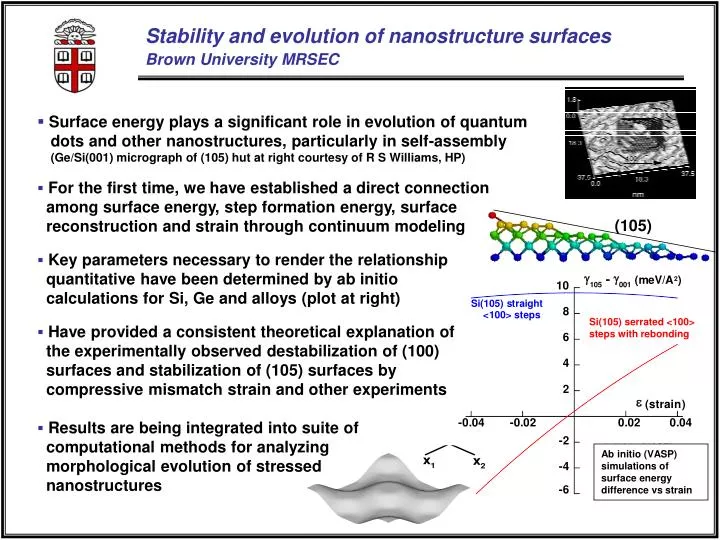
(105) Si(105) serrated <100> steps with rebonding Ab initio (VASP) simulations of surface energy difference vs strain Stability and evolution of nanostructure surfacesBrown University MRSEC Ge/Si(001) • Surface energy plays a significant role in evolution of quantum dots and other nanostructures, particularly in self-assembly (Ge/Si(001) micrograph of (105) hut at right courtesy of R S Williams, HP) • For the first time, we have established a direct connection among surface energy, step formation energy, surface reconstruction and strain through continuum modeling • Key parameters necessary to render the relationship quantitative have been determined by ab initio calculations for Si, Ge and alloys (plot at right) • Have provided a consistent theoretical explanation of the experimentally observed destabilization of (100) surfaces and stabilization of (105) surfaces by compressive mismatch strain and other experiments • Results are being integrated into suite of computational methods for analyzing morphological evolution of stressed nanostructures
Narrative: Strain and surface effects in material nanostructures The study of material nanostructures at a basic level offers the promise of major advances in the understanding of electronic, optical and/or mechanical properties of materials, as well as for the manufacture of electronic or optical devices with unprecedented performance characteristics. Strain-driven nucleation, growth and coarsening in epitaxial islands on a single crystal substrate holds promise as a means of manufacturing nanoscale devices. Progress in this area of research, particularly on the SiGe/Si material systems, provides a good illustration of the productive synergy among theory, experiment and computation that underlies progress. As early observations of islands became more refined, it was discovered that the lateral faces of the hut shaped islands in the early stages of growth were invariably (105) crystallographic surfaces, and they arose only when the mismatch strain was compressive, both features being at odds with prevailing models of surface evolution. To resolve the discrepancy, detailed calculation of the formation energies of several surface step structures on biaxially strained Si and Ge (001) surfaces were computed by both first principles methods and molecular dynamics. It was discovered that a novel rebonded surface step with a (100) orientation is strongly stabilized by compressive strain compared to all other surface reconstructions which have been proposed previously. Furthermore, the minimum energy orientation of vicinal surfaces composed of such steps was found to be a (105) crystallographic orientation. It could be concluded that the lateral faces of the hut shaped islands observed in experiments on SiGe/Si (100) faces are composed of these steps. In addition, the calculations provided an explanation for the absence of any kind of nucleation barrier for the formation of islands, as has also been recently established experimentally. Subsequently, a variational approach as a basis for studying the nonlinear dynamics of nanoscale surface undulation was developed as a basis for large-scale numerical simulations. In this way, the dependence of surface energy on strain and surface orientation can be completely determined by the features of atomistic simulation of discrete material structures and incorporated into larger scale simulations, without the need for arbitrary or ad hoc assumptions.