Download
1 / 53
540 likes | 805 Views
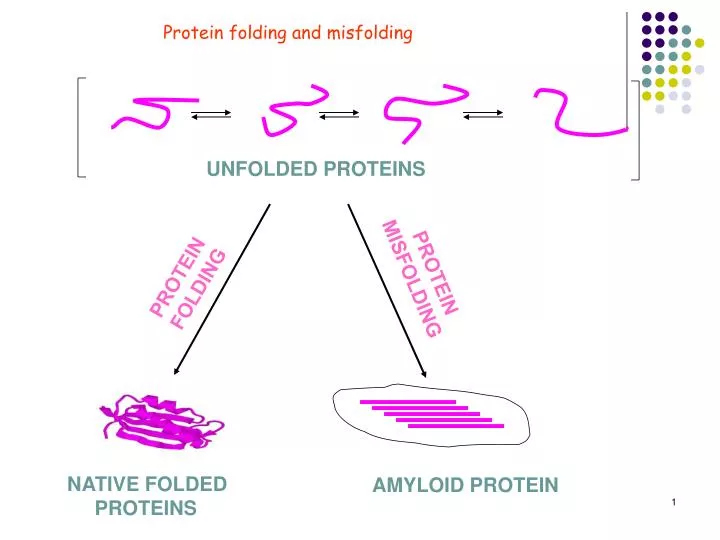
Protein folding and misfolding. UNFOLDED PROTEINS. PROTEIN MISFOLDING. PROTEIN FOLDING. NATIVE FOLDED PROTEINS. AMYLOID PROTEIN.

E N D
Protein folding and misfolding UNFOLDED PROTEINS PROTEIN MISFOLDING PROTEIN FOLDING NATIVE FOLDED PROTEINS AMYLOID PROTEIN
ll living organisms use proteins for structure, energy production, information processing, etc. Finished proteins have complicated three-dimensional structures that are necessary for their diverse activities. However, proteins are first produced as a linear string of amino acids, and they must then fold into the right conformations in the crowded environment of a cell (~300mg/ml). Diseases can ensue when proteins have problems reaching their functional conformations: sickle cell anemia, cystic fibrosis, Alzheimer’s etc.
Levinthal paradox • Assume each amino acid backbone can be in 3 conformational states, for 101 residues, there are 3100 = 5 x 1047 conformations. • If the protein can sample a new conformation at a rate of 1013 s-1, it will take 1027 years to try them all. Longer than the age of the universe! • Therefore, proteins must fold in “pre-arranged pathways” and in a cooperative manner. • Levinthal C. Extrait du Journal de Chimie Physique 1968; 65:44 Zwanzig et al., PNAS 1992; 89:20-22
Protein folding • Native state corresponds to structure that is most stable under physiological conditions • Assumption that native like interactions are on average more stable than non-native ones—search mechanism is able to find native state. • Mechanism is efficient in that only a small fraction of the conformations need to be sampled during folding
Folding Funnel A schematic energy landscape for protein folding. The surface shown here is derived from a computer simulation of the folding of a highly simplified model of a small protein. Such a surface serves to “funnel” the multitude of denatured conformations to the unique native structure. The critical region on a simple surface, such as this one is the saddle point corresponding to the transition state, the barrier that all molecules must cross to be able to fold to the native state. An ensemble of structures corresponding to the experimental transition state for the folding of a small protein is indicated in the figure; this ensemble was calculated by using computer simulations constrained by experimental data from mutational studies of acylphosphatase. The yellow spheres represent the three “key residues” in the structure; when these residues have formed their native-like contacts, the overall topology of the native fold is established. The structure of the native state is shown at the bottom of the surface, while at the top are indicated schematically some contributors to the distribution of unfolded states that represent the starting point for folding. Also indicated on the surface are highly simplified average trajectories for the folding of individual molecules. Methods. 2004,34(1):4-14.
Protein misfolding and aggregation IDP Transient dimer protofibrils Amyloid fibril Example: -synuclein (Parkinson’s), IAPP (Diabetes), A(Alzhermer’s), polyQ (Huntington’s)
Aggregation of -synuclein is related to Parkinson’s Disease • Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease in the world. • Aggregated -synuclein (aSyn) is found in the Lewy body or Lewy neurites of brain cells which is the hallmark of PD. • aSyn forms amyloid fibrils in vivo and in vitro. Lewy body Goedert M, Nat Rev Neurosci.2001,2(7):492-501. Conway KA, Biochemistry.2000,;39(10):2552-63.
Alzheimer afflicts 10 percent of those over 65 years old and perhaps half of those over 85. Every year Alzheimers not only kills 100,000 Americans, but also costs society $82.7 billion to care for its victims.In 1991, several different re- search groups found that individuals with specific mutations in their amyloid precursor protein developed Alzheimers disease as early as age 40. The body processes amyloid precursor protein into a soluble peptide (small protein) known as Aβ; under certain circumstances, Aβ then aggregates into long filaments that cannot be cleared by the bodys usual scavenger mechanisms. These aggregates then form the β-amyloid, which make up the neuritic plaque inAlzheimer patientspeptide (small protein) known as Aβ; under certain circumstances, Aβ then aggregates into long filaments that cannot be cleared by the bodys usual scavenger mechanisms. These aggregates then form the β-amyloid, which make up the neuritic plaque inAlzheimer patients.
TEM shows that all variants make fibrils T53A mouse N87S human T53A,N87S M100L,G103N,Y107A G121D, S122N Scale bar: 200 nm
Protein folding in the cell • Cellular environment is very complex—concentration is 350mg/ml. • To avoid aggregation auxilliary proteins exist to assist the proteins to fold efficiently and without aggregation • Folding catalysts to enhance slow steps in folding and molecular chaperones to avoid protein misfolding • Sophisticated mechanisms of quality control to check whether proteins are correctly folded and to destroy proteins that are not good.
Regulation of protein folding in the ER Many newly synthesized proteins are translocated into the ER, where they fold into their three-dimensional structures with the help of a series of molecular chaperones and folding catalysts (not shown). Correctly folded proteins are then transported to the Golgi complex and then delivered to the extracellular environment. However, incorrectly folded proteins are detected by a quality-control mechanism and sent along another pathway (the unfolded protein response) in which they are ubiquitinated and then degraded in the cytoplasm by proteasomes. Nature. 2003,426:884-890.
Folding in the cell • Proteins are synthesized on ribosomes in cellular DNA. Folding occurs in the cytoplasm after release from the ribosome and in other folding specific compartments such as ER. Incompletely folded proteins expose to solvent some regions that are buried in the native state and prone to inappropriate interactions with other molecules in the crowded cellular environment. Range of strategies to prevent such behavior • Molecular chaperones either interact with protein on ribosome or guide later stages of folding—chaperones work by reducing the probability of competing reactions such as aggregation • After folding in the ER secretion to the golgi apparatus. In the ER proteins must undergo quality control check—if they do not pass they are targeted for degradation • Incorrect folding—cystic fibrosis and cancer—cannot exercise function. Other proteins with high propensity to misfold form aggregates such as Alzheimers, type II diabetes and BSE or vCJD.
Protein misfolding • Misfolding is defined as reaching a state that has a significant proportion of nonnative interactions between residues • Proteins important in biological processes—failure to fold correctly—gives rise to disease • Misfolded proteins do not exercise proper function or form deposits in the cells. • Not only “loss of function” but “toxic gain of function” as aggregates induce cell damage or cell death
Protein folding and disease • Mistakes in folding will give rise to malfunctioning of biological processes and to disease. • Example cystic fibrosis—protein does not fold due to familial mutation and results in reduced level or reduction of protein. • Amyloid diseases—failure to fold results in aggregation and deposits of protein in one or more tissues.
Aggregation diseases • Give rise to deposition of proteins in the form of amyloid fibrils and plaques • Deposits in brain, organs such a liver and spleen • Fibrillar structures have similar morphology
Amyloid diseases Semin Cell Dev Biol. 2004,15(1):3-16.
Amyloid fibrils • Amyloid fibrils appear in a class of diseases called amyloid diseases such as Alzheimer’s, Parkinson’s, Huntington’s disease. • Point of contention is whether the most important toxic species is the amyloid fibril or a smaller aggregated form of the same protein or peptide. • Amyloid fibril formation is not a property that is restricted to proteins associated with disease. • Amyloid fibril appears to be a stable structural state of a polypeptide competing thermodynamically and kinetically with globular monomeric states and unfolded monomeric states.
Mechanism of protein aggregation Figure 2 A schematic representation of some of the many conformational states that can be adopted by polypeptide chains and of the means by which they can be interconverted. The transition from β-structured aggregates to amyloid fibrils can occur by addition of either monomers or protofibrils (depending on protein) to preformed β-aggregates. All of these different conformational states and their interconversions are carefully regulated in the biological environment, much as enzymes regulate all the chemistry in cells, by using machinery such as molecular chaperones, degradatory systems, and quality control processes. Many of the various states of proteins are utilized functionally by biology, including unfolded proteins and amyloid fibrils, but conformational diseases will occur when such regulatory systems fail, just as metabolic diseases occur when the regulation of chemical processes becomes impaired.
Overview of possible fates of newly synthesized chain J Mol Med. 2003;81(11):678-99.
Fate of polypeptide chain • Equilibrium 1 is affected by mutations, misprocessing, aberrant interactions with metals, changes in environmental conditions. This increases the population of partially folded or unfolded species—species are more aggregation prone than native state. • Equilibrium 2 affected by mutations so as to kinetically favor aggregate nucleation • Reaction 3 is irreversible and mature fibrils are the final stable product if aggregation process.
Common features in self assembly • First phase involves the formation of oligomeric species as a result of non specific interactions. • Smaller oligomers are soluble and earliest species observable by AFM are bead like structures. • Prefibrillar aggregates then transform into protofibrils. • Self assemble into mature fibrils.
Fig. 4. Some amyloid-related peptides/proteins form early aggregates of globular appearance that further organise into beaded chains, globular annular 'doughnut' shaped assemblies eventually giving mature protofilaments and fibrils. Pre-fibrilar aggregates may interact with reconstituted phospholipid membranes and with cell membranes where they form aspecific channels (pores) disrupting cellular homeostasis. The latter possible mechanism of toxicity is similar to that displayed by antimicrobial peptides, pore-forming eukaryotic proteins and bacterial toxins and newly synthesised cyclic peptide antibiotics (see text). The electron micrographs of the globular and beaded chains of Ab peptides are taken from Harper et al. [200]. The electron micrographs of the rings of the a-synuclein A53T (upper row) and A30P (middle row) mutants and of the Alzheimer precursor protein artic mutant (lower row) are from [201] J Mol Med. 2003;81(11):678-99.
Characterization of amyloid structures • Amyloid deposits all show characteristic optical behavior, green birefringence on binding to certain dye molecules such as Congo red because of the regularly spaced b-sheets • b-structure is seen by CD and FT-IR • EM—long and unbranched; fibrils are usually 10 nm in diameter and consist of between 2 to 6 protofibrils twisted around each other. • X-ray fibre diffraction organized core has a cross-b structure where sheets are assembled from b-strands that run perpendicular to fibrillar axis
Schematic representation of the mechanism of formation of amyloid fibrils and the various types of antibodies that can be produced. The first step in the process of formation of an amyloid fibril (black box) is the formation of an amyloidogenic intermediate via the partial unfolding of the native state (arrow 1) or via partial folding of otherwise naturally unfolded species (arrow 2). The second step is the self-association of the amyloidogenic intermediates, which eventually leads to the formation of amyloid fibrils (red box). The amyloidogenic intermediates have a high tendency to aggregate and become stabilised by the formation of intermolecular β-sheets. Small oligomers are formed initially that act as nuclei to direct the further growth of aggregates (*Here the nucleus is for simplicity shown as a dimer). This growth leads to the formation of higher order oligomers referred to as pre-fibrillar aggregates (PA, sometimes also referred to as amorphous aggregates, micelles or protofibrils). These aggregates convert into protofilaments (P) directly or indirectly, and finally into mature fibrils (F). Such fibrils usually consist of two to six protofilaments that are often twisted around each other to form a rope-like structure as shown in Box 1c. (**According to this scheme, antibodies 3 and 5 also bind to PA species but they are not represented for clarity). Biochimie. 2004;86(9-10):589-600
Fig. 3. A molecular model of an amyloid fibril. This model is derived from cryo-EM analysis of fibrils grown from an SH3 domain. The fibril consists of four “protofilaments” that twist around one another to form a hollow tube with a diameter of approximately 6 nm. The model illustrates one way in which regions of the polypeptide chain involved in β-sheet formation could be assembled within the fibrils. Semin Cell Dev Biol. 2004,15(1):3-16.
Fig. 15. Structural model for the protofilament in Aβ1–40 fibrils prepared with gentle agitation (Petkova et al. 2006), exhibiting the morphologies and NMR signals shown in Figs. 4, 8, 10a, 12–14. Residues 1–8 are conformationally disordered and are therefore omitted. The long axis of the fibril is perpendicular to the page in panels (a)–(c). The long axis is vertical and parallel to the page in panel (d). (a) All-atom representation of a pair of peptide molecules. Residues 10–22 and 30–40 have β-strand conformations, forming two separate in-register, parallel β-sheets. The protofilament is a four-layered β-sheet structure with C2 symmetry about its long axis. Blue double-headed arrows indicate side-chain–side-chain and side-chain–backbone contacts established by 2D 13C–13C NMR measurements as in Fig. 12. Purple double-headed arrows indicate contacts established by measurements of 15N–13C dipole–dipole couplings, as in Fig. 16. (b) Average structure resulting from 10 independent molecular dynamics/energy minimization runs on a cluster of 12 peptide molecules, with interatomic distance and backbone torsion angle restraints dictated by solid-state NMR data. Hydrophobic, polar, negatively charged, and positively charged side-chains are colored green, purple, red, and blue respectively. The four-layered β-sheet structure is stabilized primarily by hydrophobic interactions in the core of the protofilament. Polar and charged side-chains are on the exterior, with the exception of oppositely charged K28 and D23 side-chains, which form salt bridges. (c, d) Cartoon representations, with residues 12–21 in red and residues 30–40 in blue. A left-handed twist of 0·833°/Å is imposed, although direct experimental constraints on the twist in agitated Aβ1–40 protofilaments are not yet available. Q Rev Biophys. 2006;39(1):1-55
Figure 5 (a) Insulin structure showing the three native disulfide bonds. A chain, green; B chain, blue; disulfide bonds, gold. (b) Topology diagram of insulin color coded as in a. (c) Possible topology for the amyloid protofilament. Orientations of the termini and disulfide bonds within the curved structure are arbitrary. The C terminus of chain B (dashed) is not required for amyloid fibril formation (see ref. 43). (d) β-strand model of a protofilament. Each chain is shown in two segments, a straight and a curved β-strand (PDB accession no. 1umu, residues 93–100). Each insulin molecule would occupy two layers, connected by the interchain disulfide bonds. (e) A possible β-strand model docked into the EM density of the compact fibril (transparent gray surface). The four protofilaments are colored separately. PNAS. 2002;99(14):9196-201
Figure 6 Models for protofilament packing. (a) A twisted pair of rectangular protofilaments in which an interactive surface is colored purple. The protofilament twist accompanies the filament twist. (b) A supercoiled pair of protofilaments in which the regions involved in packing interactions rotate around each protofilament. In the correlated twist model a, interacting regions would be fixed relative to the cross-β structure, and other regions could accommodate large loops and/or folded domains that would not interfere with protofilament packing. Similar models can be constructed with more than two protofilaments, in which the cross section rotates as a rigid unit in the helical structure. Keeping the cross section fixed means that all packing contacts can be preserved in the helical fibril. This is the case for the four-protofilament model in Fig. 5e. PNAS. 2002;99(14):9196-201
Figure 1 Recent three-dimensional structural models of fibrillar aggregates from different sources. (a) The protofilament of Aβ viewed down the long axis of the fibril. The segments 12–24 (red) and 30–40 (blue) are shown. (b) The fibril from the C-terminal domain 218–289 of the fungal prion protein HET-s The ribbon diagram shows the four β-strands (orange) (residues 226–234, 237–245, 262–270, and 273–282) and the long loop between β2 and β3 from one molecule. Flanking molecules along the fibril axis (gray) are shown. (c) Atomic structure of the microcrystals assembled from the GNNQQNY peptide. Each β-strand is a peptide molecule. (d) The protofilament from amylin. Green, yellow, and pink β-strands indicate residues 12–17, 22–27, and 31–37, respectively. The unstructured N-terminal tail is shown on the right of the panel along with the disulfide bridge between Cys2 and Cys7. (e) The fibril from the NM region of Sup35p. The colored ribbons indicate residues 25–38 (red), 39–90 (blue), and 91–106 (green). The unstructured regions 1–20 (red dashed lines) and 158–250 (black dashed lines) are shown. Annu Rev Biochem. 2006;75:333-66.
In native state polypeptide chain is buried. Conditions that favor aggregation from folded proteins are those that stimulate at least partial unfolding for example low pH or elevated temperature. • Mutations that destabilize the native state are commonly involved in familial forms of amyloid disease. • Fragmentation of proteins • Formation characterized by a lag phase followed by a period of rapid growth
Determinants of protein aggregation • Thought at first that only certain proteins could form fibrils. • Ability to form amyloid fibrils is generic feature of polypeptide chain. • Fibrils formed by many proteins that are not associated with disease. • Amyloid fibrils are organized and adopted by unfolded chain when it behaves like a polymer.
Arch Biochem Biophys. 2008;469(1):100-17 Representative structures of proteins involved in disease-related amyloid fibril formation Fig. 6. Representative structures of proteins involved in disease-related amyloid fibril formation. The polypeptides are coloured according to the aggregation tendency of their amino acid sequences predicted using the algorithm TANGO. Sequences shown in blue are predicted to have no β-aggregation propensity, while polypeptide stretches coloured in yellow, orange and red indicate an increasing propensity to aggregate. Notably, the peptide structures were obtained in the presence of fluoroalcohols (calcitonin and Aβ1–42) or SDS micelles (amylin), and these sequences might be substantially less ordered in the absence of these additives. Note also that for insulin, amylin and calcitonin, the pro-peptides as well as the mature sequences have been implicated as potentially amyloidogenic.
Although the fundamental cross-b structure is common to all amyloid fibrils, the packing of b-sheets into protofilaments must vary to some extent according to the constraints of the constituent polypeptide. • Although different amyloid protofilaments appear similar in size, the protein sequence can influence the sheet packing into protofilaments
Amyloid structure • NO complete structure has been determined • Core structure is stabilized by hydrogen bonds involving the main chain. • Explains why fibrils formed from different amino acid sequences are similar in appearance. • Side chains are incorporated in manner most favorable for given sequence • Proportion of polypeptide chain that is incorporated in the core structure varies substantially—sometimes very few residues • Very different from globular proteins where highly specific packing of side chains may override main chain preferences
Questions about fibrils • Are cross b structures comprised of parallel b-sheets, antiparallel b-sheets or mixed b-sheets? • Are b-sheets structurally ordered or disordered? • Which segments of the protein sequence participate in the b-sheets? • Do amyloid fibrils really contain multiple layers of b-sheets and how are these layers arranged relative to each other? • Which aspects of amyloid structure are universal and which are sequence dependent? • What are intermolecular and intramolecular interactions that stabilize amyloid structures apart from the backbone H-bonds?
Generic nature of amyloid formation • Ability to form amyloid structures appears generic but propensity can vary dramatically between sequences • Mutation of single aa in 100 residue protein changes rate at which aggregation occurs • Change in rate can be correlated with changes in properties such as charge, secondary structure propensity and hydrophobicity (Chiti)
Importance of hydrophobicity, net charge, and secondary structure propensity • Hydrophobic interactions—important driving forces in aggregation: experiments show that residues promoting aggregation are not spread all over the sequence—instead clustered within narrow range of sequence. Different from protein folding where distant residues may nucleate folding
Charge in protein aggregation • Electrostatic effects important in modulating aggregation • Net charge is a major determinant of aggregation • Unfolded protein aggregates more rapidly when numbers of positively and negatively charged aa are equal and resulting net charge is zero
Importance of propensity to form secondary structure in aggregation • Sequences with high propensity to form b-structures are highly amyloidogenic • Hecht and co-workers designed combinatorial library of sequences that shared identical patterns of regularly alternating polar and non-polar residues ideal for b-sheet formation. Alternating periodicity is interrupted by lysine residues at positions originally occupied by non-polar aa-- amyloid formation was interrupted and sequences folded into monomeric b-sheets.
Amino acid sequences have evolved to take into account the influence of hydrophobicity, charge and b-sheet propensity • Has an evolutionary pressure existed to select against protein sequences with a high propensity to aggregate? • Folding is a strategy to escape aggregation Hydrophobics are buried Amide and carbonyl groups engaged in H-bonding Charges are exposed—promote repulsive action • Folded proteins are susceptible to aggregation Transiently adopt unfolded or partially folded conformation • Unfolded proteins: lower hydrophobic content and higher net charge contributes to maintain aggregation propensity low.
Prevention of amyloid like aggregation as a driving force of protein evolution (Montsellier and Chiti EMBO reports) • Folding is a primary strategy to prevent aggregation • Negative design to control the assembly of folded proteins: Richardsdon and Richardson have shwon that proteins have evolved structural adaptations to protect the peripheral b strands—thay rae protected by structural strategies such as covering them with a loop or a helic, formation of continuous sheet to create a beta barrel, distrotionsof b structure… • Conservation of gly and pro residues —proline have structural constraints that make it difficult to adapt the into a beta-structure. Gly have a high level of conformatioanl flexibility and high entropic cost to being incorporated into secondary structure
Use of gatekeeper residues to control aggregation: it was found that positions flanking aggregating stretches are enriched with residues such as proline, lysine, arginine, glutamate and aspartate—pro Is b-breaker and other 4 are at the bottom of the aggregation propensity scales-90% of 26,000 aggregating sequences found by TANGO have at least 1 of the 5 residues at the 1st position on either side of the segment.
Strategies for therapeutic intervention • Distinct steps in the aggregation process where intervention might be able to prevent or reverse the formation of aggregates • Destabilization of native state by genetic mutation—stabilizing native states of disease associated variants; use small molecule analogs: Example thyroxine a natural ligand of transthyretin blocks that rate at which transthyretin aggregates. • Aggregation due to fragments: peptide fragments result from natural processing or incomplete degradation of full length protein and cannot fold without the rest of the polypeptide chain. Solution: block the enzymes that generate the fragments. Alzheimers comes from 40 or 42 residue peptide that is derived from the amyloid precursor protein (APP) –secretase enzymes process APP and secretase inhibitors are under development to treat Alzheimers.
Therapeutic approaches • Enhance clearance of aggregation prone species. Active immunization with Ab peptides results in clearance of amyloid deposits in transgenic mice or a ligand that binds to serum amyloid P (SAP) a protein that associates with amyloid deposits and blocks natural clearance mechanisms reduces SAP levels • Alternative is to intervene in the aggregation process directly by small peptides or peptide analogs designed to bind to fibrils—problem is that inhibition of amyloid fibril formation might occur at the expense of the small aggregate precursors. This may be problem as small aggregates may be the primary pathogenic agents in neurodegenerative diseases. • Because of the generic nature of the amyloid formation perhaps one drug can block a number of diseases
Strategies for therapeutic intervention Therapeutic intervention in amyloid diseases. The conversion of normal soluble peptides and proteins into insoluble aggregates that are deposited in a variety of tissues is shown (5). Highlighted are stages in the aggregation process where therapeutic intervention may be able to prevent or reverse aggregation. Therapeutic strategies include (A) stabilizing the native state; (B) inhibiting enzymes that process proteins into peptides with a propensity to aggregate; (C) altering protein synthesis; (D) stimulating clearance of misfolded proteins, for example, by boosting their proteasomal degradation; (E) inhibiting fibril assembly; (F) preventing accumulation of fibril precursors. Science (2004):304.1259-1262
Folding vs. aggregation: kinetic partitioning • Amyloid fibrils are formed in a nucleation dependent manner—protein monomer is converted into a fibrillar structure via transient aggregation • Residues key to aggregation are thought to be different from those that drive the folding of polypeptide chain—although the major driving forces, hydrogen bond formation and burial of hydrophobic surface area are the same for both processes • Although a large part of the peptide is involved in fibril structure, some aa sequences are more prone to aggregation than others—this may be similar to the fact that only a few residues define the folding nucleus in protein folding.
Folding vs. aggregation Ability of proteins to fold rapidly to their globular native structure allows them to escape aberrant side reactions that would give access to the aggregation funnel and lead to thermodynamic ground state of intermolecular assembly, the amyloid fibril Evolution has shaped the folding and aggregation funnels to allow trapping of the native functional state which is thermodynamically a metastable structure in the context of the entire protein landscape. Proteins are dynamic and have transient partial unfolding events. Native proteins are marginally stable compared to denatured proteins and partially folded states can be formed from the folded structure by local and sub-global unfolding events. However, cooperativity of folding and cellular rescue machinery help to avoid populations of partially folded forms.