Download

1 / 36
360 likes | 531 Views
Transcript Complexity in the Human Genome Roderic Guig ó i Serra Centre de Regulaci ó Genòmica Barcelona. The standard model of the eukaryotic gene.

E N D
Transcript Complexity in the Human GenomeRoderic Guigó i SerraCentre de Regulació GenòmicaBarcelona CSHL Programming for Biology, fall 2008
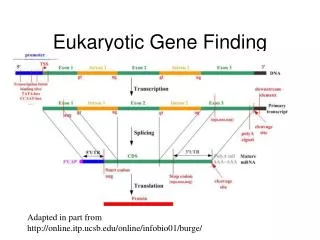
The standard model of the eukaryotic gene most of the transcriptional output of the human genome is localized in well defined genomic loci, which occupy a small fraction of the genomic space, and which encode mRNAs that, when exported into the cytosol, are translated into proteins CSHL Programming for Biology, fall 2008
cDNA cloning and sequencing IBM Systems Journal, Inman et al (2001) , Deep Computing for life Sciences CSHL Programming for Biology, fall 2008
5 4 3 1 2 11 8 9 10 7 12 6 18 16 17 15 14 13 20 21 22 Y 19 X CSHL Programming for Biology, fall 2008
The GENCODE reference gene set (as part of the ENCODE project) • mapping of known transcripts sequences (ESTs, cDNAs, proteins) into the human genome • manual curation to resolve conflicting evidence • additional computational predictions • experimental verification • FINAL ANNOTATION CSHL Programming for Biology, fall 2008
The GENCODE reference gene set (as part of the ENCODE project) • mapping of known transcripts sequences (ESTs, cDNAs, proteins) into the human genome • manual curation to resolve conflicting evidence • additional computational predictions • experimental verification • FINAL ANNOTATION CSHL Programming for Biology, fall 2008
The gencode pipelinemanual curation: havana (sanger)experimental verification:genevabioinformatics: imim • 2608 transcripts in 487 loci • 137 transcripts in 53 non-coding loci • 1097 coding transcripts and 1374 non-coding transcripts in 434 protein coding loci • most of protein coding loci encode • a mixture of protein coding and • non-coding transcripts
CSHL Programming for Biology, fall 2008
EGASP’05 • Automatic methods were not able to complete reproduce the GENCODE manua annotation • Although some performed remarkably well • Very few novel computational predictions could be verified experimentally • Suggesting completeness of GENCODE CSHL Programming for Biology, fall 2008
one gene - many proteinsvery complex transcription units CSHL Programming for Biology, fall 2008
high dynamic range in RNA abundance • RNA species vary greatly in abundance. Typically (Carninci et al., 2000) • 5-10 highly expressed species. thousands of copies ~ 20% of the mRNA mass • 500-2000 intermediate species. hundreds of copies 40%-60% of the mRNA mass • 10,000-20,000 rare messages. a few copies<20%-40% of the mRNA mass • Random clone selection is ineffective in recovering low abundance transcripts. CSHL Programming for Biology, fall 2008
Unanticipated Transcript complexity Many individual studies suggest unanticipated complexity of the transcriptional map of the human genome: • Kapranov et al. (2007)RNA onto tiling arrays, novel RNA classes, hundreds of thousands of novel sites of transcription • Peters et al. (2007)LongSage, evidence for thousands of novel transcripts • Roma et al. (in press)gene trap sequence tags in mouse embryonic stem cells, thousands of novel transcripts • Unneberg and Claverie (2007)interchromosomal transcript chimerism • Denoeud et al. (2007)RACEarrays. Doubling the number of annotated exons in protein coding transcripts, widespread transcript chimerism CSHL Programming for Biology, fall 2008
Genome tiling arrays Slide from http://signal.salk.edu/msample.html Salk Institute Genomic Analysis Laboratory CSHL Programming for Biology, fall 2008
TRANSCRIPTION OF PROCESSED POLY A+ RNA based on a number of high throughput technologies within the ENCODE project Fraction of Bases in Primary Transcripts92% CSHL Programming for Biology, fall 2008
tiling arrays reveal many novel sites of transcription TRANSCRIPTION MAP of HL-60 DEVELOPMENTAL TIME COURSE (data by Tom Gingeras, affymerix) CSHL Programming for Biology, fall 2008
If we select 40 clones at random from the RACE reaction, the probability of selecting a clone from the less abundant form is 0.01 (assuming a multinomial distribution) CSHL Programming for Biology, fall 2008
RACEarrays: an strategy for normalization of RACE libraries, and exhaustive identification of alternative transcripts CSHL Programming for Biology, fall 2008
Array based normalization of RACE libraries If we select 40 clones at random from the RACE reaction, the probability of selecting a clone from the less abundant form is 0.01 (assuming a multinomial distribution) However, if the transcript forms could be segregated by RT-PCR, then by selecting again 40 random clones, 10 from each RT-PCR, the probability of selecting the less abundant form is now, 0.6 CSHL Programming for Biology, fall 2008
MECP2 locus:2 known transcript variants (4 known exons)15 new variants discovered (14 new exons) CSHL Programming for Biology, fall 2008
Experiments on chr21 and chr22 • 492 protein coding genes interrogated • 1,668 exons selected for 3’ and 5’ RACE • RACE performed in 16 tissues and cell lines • RACE reactions pooled before hybridization into chr21 and chr22 Affymetrix tiling arrays • 102 pools • Pooling designed to maximize the distance between consecutive primers within the same pool (to facilitate assignment of RACEfrags to the RACE index exons) CSHL Programming for Biology, fall 2008
RACEfrags on chr21 and chr22 CSHL Programming for Biology, fall 2008
Characterization by sequencing CSHL Programming for Biology, fall 2008
Distal transcript connections A 5.4 Mb canonical “intron” confirmed by RT-PCR and sequencing CSHL Programming for Biology, fall 2008
There is concordance between transcriptional networks in different tissues (ch22) CSHL Programming for Biology, fall 2008
Lévy Flight distribution? Distribution of distances RACEfrag to index exon CSHL Programming for Biology, fall 2008
summary • There is a lot of transcriptional activity , which appears to be of unexpected complexity, and it has been largely unexplored • Although instruments need to be calibrated CSHL Programming for Biology, fall 2008
WHAT ARE ALL THESE TRANSCRIPTS DOING? CSHL Programming for Biology, fall 2008
The GENCODE annotation • 487 loci. 2608 transcripts • 53 non-coding loci. 137 transcripts • 434 protein coding loci. • 1097 coding transcripts • 1374 non-coding transcripts • 5.7 transcripts per protein coding locus • 2.5 coding transcripts per locus • 1.7 proteins per locus CSHL Programming for Biology, fall 2008
the combined analysis of BioSapiens, Kellis and Goldman identified 184 annotated protein coding transcripts which challenged (from the structural, functional and/or evolutionary standpoint) our current view of proteins. Footnote: removing these 184 proteins from the set of 738 GENCODE proteins, will leave 554 proteins for 434 loci; barely 1,3 proteins per locus CSHL Programming for Biology, fall 2008
Structural Effects of Pepsinogen C Alternative Splice Variant Locus RP11-298J23.1 codes for pepsinogen C. The structure of pepsinogen C is 1htrA. Isoform -003 is missing 80 residues with respect to pepsinogen C. Here the missing section of -003 is in light green. The missing section in this isoform would remove the core from both subdomains of the structure. Both the N-terminal sub-domain (on the left) and the C-terminal sub-domain would have to refold. This is the view from above looking down into the active cleft of the proteinase. Active site aspartates are shown in ball and chain. One of the two active site residues is in the missing section. The symmetry apparent in this isoform suggests that although it will have to refold it may very well be able to reform into a single subdomain. Michael Tress & Alfonso Valencia CNB, Madrid
Proving translation to protein • Q-PCR on the canonical and alternative forms to measure transcription levels (Stylianos Antonarakis, University of Geneve) Are the expression levels of the alternative form comparable to those of the constitutive form? • RT-PCR on polysomal RNAs of the canonical and alternative form (Stylianos Antonarakis, University of Geneve) Is the alternative form bound to the ribosome? • Camel antibodies against the alternative splice junction (Sylvere van der Maarel) Is the alternative form translated to protein? • Measurement of the half life of the canonical and alternative form (Alex Reymond, University of Lausanne)Is the alternative translated protein stable? • pattern of protein interactions in the canonical and the alternative forms (Kouros Salehi-Ashtiani, Dana Farber Cancer Institute)Is there evidence of differential functionality for the alternative protein?
Expression levelsalternative vs constitutive • Q-PCR in three cell lines: • SKNAS • GM06990 • HelaS3 CSHL Programming for Biology, fall 2008
Polysomal associationalternative vs constitutive CSHL Programming for Biology, fall 2008
Center for Genomic Regulation Roderic Guigó Julien Lagarde France Denoeud (in France) Sarah Djebali Sylvain Foissac (in Affymetrix) Vincent Lacroix David Martin Micha Sammet Paolo Ribeca University of Geneva Stylianos Antonarakis Catherine Ucla Sam Deutsch University of Lausanne Alex Reymond Cedric Howald The Sanger Institute Tim Hubbard Jen Harrow Adam Frankish Affymetrix Tom Gingeras Phil Kapranov Jorg Drenkow Ian Bell Erica Dumais Dana-Farber Kourosh Salehi-Ashtiani Ryan R Murray Chenwei Lin David Szeto Marc Vidal Université du Quebec Ann Bergeron CNIO Alfonso Valencia Michael Tress
CENTRE DE REGULACIÓ GENÒMICA PARC DE RECERCA BIOMÈDICA DE BARCELONA CSHL Programming for Biology, fall 2008