Download

1 / 63
660 likes | 1.11k Views
The Organic Chemistry of Enzyme-Catalyzed Reactions Revised Edition. Professor Richard B. Silverman Department of Chemistry Department of Biochemistry, Molecular Biology, and Cell Biology Northwestern University.

E N D
The Organic Chemistry of Enzyme-Catalyzed ReactionsRevised Edition Professor Richard B. Silverman Department of Chemistry Department of Biochemistry, Molecular Biology, and Cell Biology Northwestern University
The Organic Chemistry of Enzyme-Catalyzed ReactionsChapter 1Enzymes as Catalysts
For published data regarding any enzyme see: http://www.brenda-enzymes.info/
First “isolation” of an enzyme in 1833 • Ethanol added to aqueous extract of malt • Yielded heat-labile precipitate that was utilized to hydrolyze starch to soluble sugar; precipitate now known as amylase • 1878 - Kühne coined term enzyme - means “in yeast” • 1898 - Duclaux proposed all enzymes should have suffix “ase” What are enzymes, and how do they work?
Enzymes - natural proteins that catalyze chemical reactions • First enzyme recognized as protein was jack bean urease • Crystallized in 1926 • Took 70 more years (1995), though, to obtain its crystal structure
Enzymes have molecular weights of several thousand to several million, yet catalyze transformations on molecules as small as carbon dioxide and nitrogen • Function by lowering transition-state energies and energetic intermediates and by raising the ground-state energy • Many different hypotheses proposed for how enzymes catalyze reactions • Common link of hypotheses: enzyme-catalyzed reaction always initiated by the formation of an enzyme-substrate (or ES) complex in a small cavity called the active site
1894 - Lock-and-key hypothesis - Fischer proposed enzyme is the lock into which the substrate (the key) fits • Does not rationalize certain observed phenomena: Compounds having less bulky substituents often fail to be substrates Some compounds with more bulky substituents bind more tightly Some enzymes that catalyze reactions between two substrates do not bind one substrate until the other one is bound
1958 - Induced-fit hypothesis proposed by Koshland: When a substrate begins to bind to an enzyme, interactions induce a conformational change in the enzyme Results in a change of the enzyme from a low catalytic form to a high catalytic form Induced-fit hypothesis requires a flexible active site
Concept of flexible active site stated earlier by Pauling (1946): Hypothesized that an enzyme is a flexible template that is most complementary to substrates at the transition state rather than at the ground state Therefore, the substrate does not bind most effectively in the ES complex As reaction proceeds, enzyme conforms better to the transition-state structure Transition-state stabilization results in rate enhancement
Only a dozen or so amino acid residues may make up the active site • Only two or three may be involved directly in substrate binding and/or catalysis
Why is it necessary for enzymes to be so large? • Most effective binding of substrate results from close packing of atoms within protein • Remainder of enzyme outside active site is required to maintain integrity of the active site • May serve to channel the substrate into the active site Active site aligns the orbitals of substrates and catalytic groups on the enzyme optimally for conversion to the transition-state structure-- called orbital steering
Enzyme catalysis characterized by two features: specificity and rate acceleration • Active site contains amino acid residues and cofactors that are responsible for the above features • Cofactor, also called a coenzyme, is an organic molecule or metal ion that is essential for the catalytic action
Two types of specificity: (1) Specificity of binding and (2) specificity of reaction Specificity of Binding • Enzyme catalysis is initiated by interaction between enzyme and substrate (ES complex) • k1, also referred to as kon, is rate constant for formation of the ES complex • k-1, also referred to as koff, is rate constant for breakdown of the complex • Stability of ES complex is related to affinity of the substrate for the enzyme as measured by Ks, dissociation constant for the ES complex Specificity of Enzyme-Catalyzed Reactions
Generalized enzyme-catalyzed reaction kon Michaelis complex koff Scheme 1.1 When k2 << k-1, k2 called kcat (turnover number) Ks called Km (Michaelis-Menten constant) kcat represents the maximum number of substrate molecules converted to product molecules per active site per unit of time; called turnover number
Km is the concentration of substrate that produces half the maximum rate • Km is a dissociation constant, so the smaller the Km the stronger the interaction between E and S • kcat/Km is the specificity constant - used to rank an enzyme according to how good it is with different substrates kcat Upper limit for is rate of diffusion (109 M-1s-1) Km
How does an enzyme release product so efficiently given that the enzyme binds the transition state structure about 1012 times more tightly than it binds the substrate or products? After bond breaking (or making) at transition state, interactions that match in the transition-state stabilizing complex are no longer present. Therefore products are poorly bound, resulting in expulsion. As bonds are broken/made, changes in electronic distribution can occur, generating a repulsive interaction, leading to expulsion of products
E • S complex Figure 1.1
∆Gº = -RTlnKeq If Keq = 0.01, ∆Gº of -5.5 kcal/mol needed to shift Keq to 100
Specific Forces Involved in E•S Complex Formation Examples of ionic, ion-dipole, and dipole-dipole interactions. The wavy line represents the enzyme active site Figure 1.2
Hydrogen bonding in the secondary structure of proteins: -helix and -sheet. H-bonds H-bonds A type of dipole-dipole interaction between X-H and Y: (N, O) Figure 1.3
When a molecule (or group) that is a good electron donor comes into contact with a molecule (or group) that is a good electron acceptor, donor may transfer some of its charge to the acceptor Charge Transfer Complexes
When two nonpolar groups, each surrounded by water molecules, approach each other, the water molecules become disordered in an attempt to associate with the water molecules of the approaching group • Increases entropy, resulting in decrease in the free energy (G = H-TS) Hydrophobic Interactions
Atoms have a temporary nonsymmetrical distribution of electron density resulting in generation of a temporary dipole • Temporary dipoles of one molecule induce opposite dipoles in the approaching molecule van der Waals Forces
Can be absolute or can be very broad • Specificity of racemates may involve E•S complex formation with only one enantiomer or E•S complex formation with both enantiomers, but only one is converted to product • Enzymes accomplish this because they are chiral molecules (mammalian enzymes consist of only L-amino acids) Binding Specificity
Resolution of a racemic mixture Binding specificity of enantiomers diastereomers Scheme 1.2
Binding energy for E•S complex formation with one enantiomer may be much higher than that with the other enantiomer • Both E•S complexes may form, but only one E•S complex may lead to product formation • Enantiomer that does not turn over is said to undergo nonproductivebinding
Basis for enantioselectivity in enzymes Steric hindrance to binding of enantiomers Leu S R Figure 1.4
Unlike reactions in solution, enzymes can show specificity for chemically identical protons Reaction Specificity
Enzyme specificity for chemically identical protons. R and R on the enzyme are groups that interact specifically with R and R, respectively, on the substrate. Figure 1.5
An enzyme has numerous opportunities to invoke catalysis: • Stabilization of the transition state • Destabilization of the E•S complex • Destabilization of intermediates • Because of these opportunities, multiple steps may be involved Rate Acceleration
Effect of (A) a chemical catalyst and (B) an enzyme on activation energy Figure 1.6 1010-1014 fold typically
Enzyme catalysis does not alter the equilibrium of a reversible reaction; it accelerates attainment of the equilibrium
Rate enhancement by proximity • Enzyme serves as a template to bind the substrates • Reaction of enzyme-bound substrates becomes first order • Equivalent to increasing the concentration of the reacting groups • Exemplified with nonenzymatic model studies Mechanisms of Enzyme CatalysisApproximation
Second-order reaction of acetate with aryl acetate Scheme 1.3
Covalent Catalysis Nucleophilic catalysis anchimeric assistance Scheme 1.4 Most common Cys (SH) Ser (OH) His (imidazole) Lys (NH2) Asp/Glu (COO-)
Anchimeric assistance by a neighboring group Scheme 1.5
Early evidence to support covalent catalysis Model Reaction for Covalent Catalysis Scheme 1.6
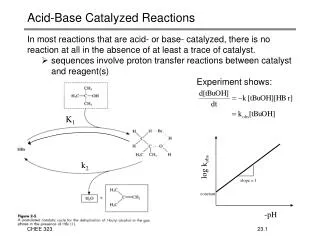
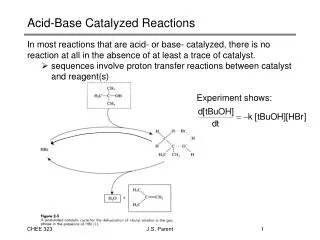
General Acid/Base Catalysis This is important for any reaction in which proton transfer occurs
The catalytic triad of -chymotrypsin. The distances are as follows: d1 = 2.82 Å; d2 = 2.61 Å; d3 = 2.76 Å. catalytic triad Figure 1.7
Charge relay system for activation of an active-site serine residue in -chymotrypsin Scheme 1.7
pKa values of amino acid side-chain groups within the active site of enzymes can be quite different from those in solution • Partly result of low polarity inside of proteins Molecular dynamics simulations show interiors of these proteins have dielectric constants of about 2-3 (dielectric constant for benzene or dioxane) • If a carboxylic acid is in a nonpolar region, pKa will rise • Glutamate-35 in the lysozyme-glycolchitin complex has a pKa of 8.2; pKa in solution is 4.5 • If the carboxylate ion forms salt bridge, it is stabilized and has a lower pKa
Basic group in a nonpolar environment has a lower pKa • pKa of a base will fall if adjacent to other bases • Active-site lysine in acetoacetate decarboxylase has a pKa of 5.9 (pKa in solution is 10.5)
Two kinds of acid/base catalysis: • Specific acid or specific base catalysis - catalysis by a hydronium (H3O+) or hydroxide (HO-) ion, and is determined only by the pH • General acid/base catalysis - reaction rate increases with increasing buffer concentration at a constant pH and ionic strength
Effect of the buffer concentration on (A) specific acid/base catalysis and (B) general acid/base catalysis Specific acid/base catalysis General acid/base catalysis Figure 1.8
Specific Acid-Base Catalysis Hydrolysis of ethyl acetate Scheme 1.8
Alkaline hydrolysis of ethyl acetate Scheme 1.9
Acid hydrolysis of ethyl acetate Scheme 1.10