Download
1 / 7
70 likes | 343 Views
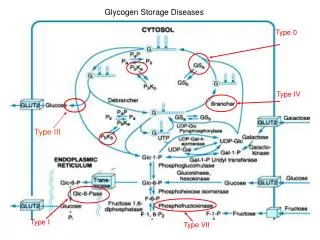
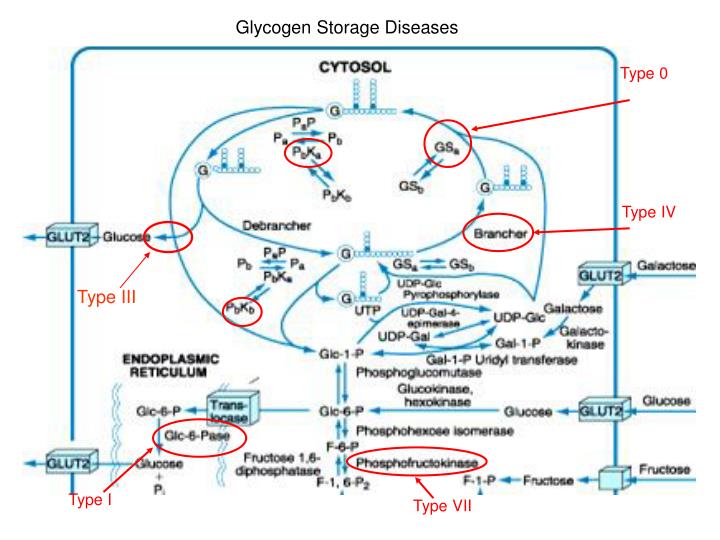
Glycogen Storage Diseases. Type 0. Type IV. Type III. Type I. Type VII. Excess glucose. Storage. Glycogen. Starch. Vertebrates Microorganisms. Plants.

E N D
Glycogen Storage Diseases Type 0 Type IV Type III Type I Type VII
Excess glucose Storage Glycogen Starch Vertebrates Microorganisms Plants Glycogen granules -- complex aggregates of glycogen and the enzymes that synthesize it and degrade it, and the machinery for regulating these enzymes.
Glycogenin (glucosyltransferase activity) Glycogenin (chain extending activity) Glucose 6-phosphate Glycogen synthase Glucose 6-phosphatase Glycogen branching enzyme 1 3 5 6 4
Type І GSD (von Gierkes disease):----- The most common and severe form of glycogen storage disease. Patients with Type І GSD are unable to release glucose from glycogen due to the deficiency of glucose-6- phosphatase and hence with time glycogen builds up in the liver. ----- Characterized by massive enlargement of liver (hepatomegaly), growth retardation, fasting hypoglycemia, increased lactic acid concentrations in the blood (due to excessive glycolysis), hyperuricemia and hypertriglyceridemia. ----- Treatment consists of providing frequent meals and naso-gastric feeding at night to maintain blood glucose concentration. Type Іb ----- has been identified as a defect in the glucose–6–phosphatase transport system. ----These patients in addition to the problems described above develop frequent bacterial and fungal infection, due to abnormal functioning of the white blood cells. Type II Type II GSD affects predominantly the heart and skeletal muscle producing muscle weakness and cardiomegaly. Liver function is normal and patients do not have hypoglycemia. ----- caused by a lack of function of the enzyme acid glucosidase, which is present in lysosomes. Without the proper functioning of this enzyme, the glycogen that comes into the lysosomes is not broken down, but accumulates and disrupts the normal functions of the cell. In muscle tissue, these enlarged lysosomes eventually cause the cells to become dysfunctional and die. There are mainly two forms of Type II GSD. ------ Infantile form (Pompe’s disease): This appears in the first few months of life with weakness and respiratory difficulties. The patients usually die before 12 months of age due to cardiac failure and respiratory weakness. Juvenile forms: A milder form and may present in the second or third decade of life with muscle weakness and difficulty in walking. ------ Treatment of aimed at relieving stress on the muscles. A protein-rich diet is used, along with an intensive daily exercise program.
Type III Deficiency of Amylo-1, 6–Glucosidase the debranching enzyme results in storage of an abnormal form of glycogen. Two subtypes of this disorder IIIa & IIIb have been observed. In GSD Type IIIa, the disease involves both liver and muscle tissues, producing hepatomegaly and muscle weakness. Type IIIb involves only the liver without apparent muscle disease. Clinical and biochemical features resemble those of type І disease. Differentiation from type І is by a lower concentrations of urate and lactate in the blood and elevated serum transaminase and creatinine kinase activities. Treatment of Type III GSD consists of frequent feedings and a high protein diet. Continuous nasogostric feedings similar to those used for Type І GSD are useful. Type V In the absence of phosphorylase in muscles, glucose cannot be released from the glycogen stored in skeletal muscles for energy. Hence people with Type V GSD, also called McArdle disease, experience problems performing and completing most exercises. These patients experience muscle pain, muscle cramps, muscle fatigue and muscle tenderness. With the breakdown of muscle and the release of myoglobin, myoglobinuria may develop. These patients should exercise moderately, for extensive exercise can cause considerable muscle breakdown resulting in great deal of release of myoglobin in the urine. However, patients with type V GSD respond to oral glucose administration. Type VI Type VI GSD also known as Hers’ disease is a rare and relatively benign disorder due to the deficiency of liver phosphorylase or one of the subunits of phosphorylase kinase. Enlargement of liver due to increased deposit of glycogen, growth retardation and mild hypoglycemia are seen.
Type VII Patients with Type VII GSD also known as Tarui’s disease have deposits of abnormal glycogen in muscle. With the deficiency of phosphofructokinase, effective glycogen breakdown (glycolysis) during muscle stress cannot be accomplished, resulting in pain, weakness, and cramping in the exercising muscle. Exercise intolerance, unresponsiveness of glucose administration and decreased glycolysis in erythrocytes produces myoglobinuria, hyperbilirubinemia and reticulocytosis.