Download

1 / 38
380 likes | 513 Views
Glycosaminoglycans and ocular structure. Class 8 Dr. Pittler. PHOTORECEPTOR LIPIDS. Photoreceptors transduce light energy into a neuro- electrical signal that is sent to area 17 of the brain which is perceived as light. The early part of transduction , involving rhodopsin and its

E N D
Glycosaminoglycans and ocular structure Class 8 Dr. Pittler
PHOTORECEPTOR LIPIDS Photoreceptors transduce light energy into a neuro- electrical signal that is sent to area 17 of the brain which is perceived as light. The early part of transduction, involving rhodopsin and its G-protein (transducin), requires the presence of a highly fluid membrane in the disks or disk- like structures.
The way in which nature has seen fit to give sufficient flexibility to photoreceptor membranes is to insert significant quantities of cervonic acid into the disk and disk-like membranes. This fatty acid has 6 double bonds and can nearly twist backwards upon itself. This places considerable disorder into the membranes into which it is inserted as a phospholipid. From the table it can be seen that marked percentages of 22:6 exist in three of the phospholipids. CERVONIC ACID (dicosahexaenoic acid) 22:6D4,7,10,13,16,19
Cervonic acid is synthesized from lenolenic acid (18:3) and this precursor must be obtained from the diet. The general pathway (through many steps) is: 18:3 ---------------> 22:5 -------------->24:6 -----------------> 22:6 linolenate docosapentate tetracosahexate cervonate Synthesis takes place in the liver, and after transport, the very long chain fatty acid is transported to the inner segment of the photoreceptors. There it is incorporated into phospho- lipids. Since this fatty acid is vital to visual transduction, there exists a “sparing” effect for it in the photoreceptors. As you will see, it is preserved after removal from photoreceptor membranes and taken back up again into photoreceptor inner segments.
time Due to the high metabolic rate of photoreceptors, the disks of the outer segments are replaced about every ten days. The turnover of disk membranes in photoreceptors was demonstrated over 20 yrs ago by R. Young using radiolabelled membrane precursors. In a series of experiments, it is possible to see the initial precursor assembly at the inner segment (1 and 2), incorporation into disk membranes (3), transport to the distal end of the rod outer segment (3->4), and shedding to a PE organelle (4->5).
At this point (incorporation into PE organelles), there is a choice • to be made regarding the disk components including the long • chain, unsaturated fatty acids such as cervonic acid. It is already • known that sparing (i.e., re-use) of both opsin and vitamin A • occurs. • It was not known, at first, what occurred with the highly unsaturated • fatty acids that are vulnerable to oxidation of their double bonds • and destruction of the acid into shorter chain aldehydes. Logically, • there is good reason to naturally preserve and re-use as many of • the fatty acids as possible since: • their synthesis is limited and metabolically complicated; • the retina has a significant supply of anti-oxidants (vitamin E)
With that in mind, investigators decided to follow the transport and sparing of [3 H] 22:6n3, a tritiated form of cervonic acid, in its progression through the neural retina using frog retinas as an animal model. The figure on the right shows the incorporation as radioactive black dots. After 4 hours, uptake can be seen in the inner segment (green arrow) of one type of rod cell (502). However, the 435-rods had already taken up cervonic acid into their outer segments (red arrow). There was no uptake into cone cells by this period (as shown by the black asterisk) which reflects the typical slower turnover of cone cells.
The data compare the uptake of radiolabelled cervonic acid in the outer segments (top graph) with that in the inner segments (lower graph). By comparing time and grain density, it can be seen that incorporation in the inner segments precedes incorporation in the outer segments. By 6 hr, incorporation had not been made to outer segment rods, but was seen in the inner segments. Although not shown here, data also showed a bidirectional labelling (at the inner segments that suggested sparing of cervonic acid).
Cervonic acid turnover in the retina. Dietary 18:3 is incorporated into the liver where 22:6PL is synthesized and transported to the photoreceptor inner segments. 22:6PL is incorporated into outer segment disks (or disk-like membranes) and removed with disk shedding. The 22:6PL is returned to the inner segment via the interphotoreceptor matrix or to the liver for re-transport to the retina.
PHOTORECEPTOR LIPIDS SUMMARY & STUDY GUIDE • 1. Why is it important to have highly unsaturated fatty acids in disks and disk-like • membranes of photoreceptors? [One word is not an answer] • 2. How would you explain what cervonic acid is? • 3. What is meant by the “sparing” effect for cervonic acid? Why is it important? • 4. Describe an experiment that shows the incorporation of cervonic acid into • photoreceptors. Further explain how the same experiment could be used to • show the sparing effect. • 5. Can you diagram cervonic acid turnover with enough detail to make it meaningful?
OCULAR GLYCOSAMINOGLYCANS USEFUL & USELESS
What we are going to consider in this lecture: • Basic structural properties of GAGs and their • functional properties in tissues. • How GAGs associate with glycoproteins • How holo-glycoproteins (proteins + GAGs) are • formed and assembled in the eye (cornea & • vitreous • 4. GAG pathology (general and ocular)
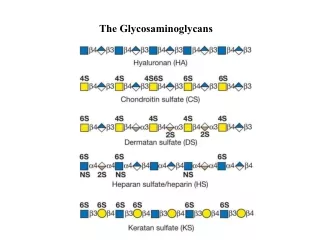
The connective, extracellular tissue that we find in the eye has two sides: the collagen side and the glycosaminoglycan side – both must work together to form tissues. Glycosaminoglycans (GAGs) were formerly called mucopolysaccharides since they were originally discovered in mucous or mucoid material such as nasal discharges and around sensitive areas of body openings. From that term we have the diseases that are known as mucopolysaccharidoses, a term that is still used today. GAGs are polymers that consist of repeating two sugar units of a sugar and an amino sugar.
An example unit is shown here: 6 5 4 1 3 2 20-60 stands for the number of two sugar units found in this GAG • NOTE: • the N-acetylated group in the right hand sugar • the presence of negative charges on the carboxylate and sulfate groups • the alternating beta 1-> 3 and beta 1-> 4 linkages • a carboxylate in the position is uronate; in the position is an iduronate • sulfation make take place in several positions – each adds another negative charge
Here are four basic GAG units that occur in the eye. Hyaluronic acid (hyaluronate) -- a component of the vitreous-- has one less charge per unit and contains N-acetyl glucose rather than N-acetyl galactose. Keratan sulfate -- a component of the cornea, like CS -- has galactose in the left hand unit, but the N-glucosamine in the right hand unit. Dermatan sulfate – also a component of the cornea – contain iduronic acid, but is otherwise like chondrointin sulfate. These small differences do not seem to make much difference in the corneal components except for the accumulation of negative charges. One other point is that the corneal GAGs have relatively short lengths (~120 glycan units) whereas hyaluronate is composed of ~50,000 glycan units per molecule. It can, therefore, be contained in a much larger volume.
There are six glycan units in this partial structue of hyaluronate. If you can imagine a molecule with 50,000 of these units, then you would have an idea of what one hyaluronate molecule looks like in the vitreous. The point is that these molecules (in very twisted and curved forms) absorb tremendous volumes of water and help the gel to have viscoelastic properties. A viscoelastic property allows deformation with the ability to return to an original shape with the same volume.
GAGs ARE BOUND TO GLYCOPROTEIN APOPROTEINS This may sound like double-talk, but it is inherited with the difficulty that arose from the literature over the years. Here is the general classification: GLYCOPROTEIN (a protein to which sugars are bound) GLYCOPROTEINPROTEOGLYCAN (has bound oligosaccharides) (has GAGs bound to it) Here’s the difficulty: a proteoglycan may refer to the holoprotein or the apoprotein (protein less the GAGs) and the literature is not always clear in making the distinction. HERE we will always refer to the holoprotein. Also glycoprotein may be general or refer to a protein bound to oligosaccharides.
GLYCOPROTEIN EXAMPLE: Rhodopsin PROTEOGLYCAN EXAMPLE: Lumican LINK OLIGOSACCHARIDE: GAL-GAL-XYLOSE (-Ser-PROTEIN)
CORNEAL PROTEOGLYCANS There are two proteoglycans in the human corneal stroma: decorin and lumican. Each protein has a molecular weight of ~40,000 D and can bind 1-3 GAGs. Strangely enough, these proteoglycans are also glycoproteins – in the sense that there is an oligosaccharide at one end of the molecule and a “link” oligosaccharide that connects the protein with each GAG. Lumican binds only keratan sulfate while decorin may bind to either chondroitin sulfate, dermatan sulfate or keratan sulfate. These molecules act as molecular spacers between type I collagen fibers and also exhibit some viscoelasticity.
Shown here are two collagen tissue sections from the corneal stroma (on the left) and the sclera (on the right). The illustrations indicate that the fibers are separated, even though the separation of scleral fibers is less organized. So spacing is a characteristic of fiber separation in both tissues.
This is an immunoelectron micrograph using a labelled (dark splotches) antibody to decorin in the human sclera.
Collagen types V/VI seem to have two roles: they limit type I fiber diameters and they connect type I collagen with proteoglycans. Proteoglycan “fibers” are shown at right angles to the collagen type I fibers in A (see red arrows). The fibers were stained with copper blue and MgCl2. The diagram in B represents how GAG- proteoglycans act as spacers between collagen fibrils. Collagens types V/VI are the “go between” molecules that connect type I fibers with the proteoglycans (not shown in B).
CURRENTLY, cross-sectional areas of the corneal stroma (within a lamella) have been proposed to have the geometrical pattern shown on the right. Each collagen fiber is shown in green. The proteoglycans are indicated as six lines radiating from each fiber by blue lines. The water between each fiber is indicated by light purple coloring. This is suggested as being the best possible model to maintain equidistances from one fiber to the next.
GAGs IN THE VITREOUS -- The GAG in the vitreous is hyaluronate. It is not associated with a proteoglycan apoprotein, but it does seems to be bound to the type IX/XI connecting collagen that surrounds the main type II collagen found in the vitreous. The drawing on the right is the general pattern of type II collagen fibers in the vitreous and indicates that GAGs (namely hyaluronate) is sandwiched in between the fibers.
This is an enlarged segment of vitreous and shows better details of the relationship between collagen and GAGs. Note that the collagen fibers are type II, the fibrils are the type IX/XI hybrid collagen, and the hyalu- ronate is depicated as “spaghetti” placed between the collagen. The hyaluronate contains large volumes of water and swells the two components. This gel is both elastic and readily transmits the IOP. (hybrid type IX/XI “FACIT” collagen) (type II)
Here is a better view of type II collagen in the vitreous (A&B). Coming out from the fibers are thin lines of type IX/XI collagen (red arrow). A diagram of that collagen is seen below the figure. This collagen has areas that are non-collagenous (FACIT collagen = fibril associated collagen with interrupted triple helices) and are designated “NC”. In the type IX/XI hybrid, NC4 is considered to be the non- collagenous area that associates with hyaluronate. As can be seen the organization of collagen and GAG are much less organized than in the corneal stroma. Yellow arrows indicate possible NCs.
USELESS GLYCOSAMINOGLYCANS….WHERE DO OLD GAGs GO WHEN THEY DIE? Generally, when molecules turnover, due to partial breakdown and loss of funtion, they are transported to lysosomes where low pH and a host of about 30 degradative enzymes will convert them to simpler molecules that can be reused. Problems arise, however, when for a genetic or other reason some of the degradative enzymes are either missing or non- functional. This has also been seen, for example in metabolism in the case of galactosemia where one of three enzymes may be non functional. Sometimes the enzymes are made, but due to signalling defects the enzymes fail to be transported to the lysozymes and may even be mistakenly moved out of the cell.
When glycosaminoglycans run into this problem, the disease that results from it are called mucopolysaccharidoses. All of these diseases are rare and all of them involve defects of degradative enzymes. It is often the case that the GAGs are only partially broken down and the process halts when the step involving the defective enzyme is encountered. These diseases often involve the eye, particularly the cornea and/or the retina. The pathology occurs due to the fact that the partly degraded GAGs accumulate in the lysosomes, engorge the cells and then spill out into nearby tissues. This situation is particularly devastating to limbs and joints as well as brain tissues. If the disease begins at birth (and often does) then mental retardation quickly establishes itself. Diseases of this type are also known as metabolic storage diseases and can involve lipids and well as polysaccharides.
FREQUENCY Each lysosomal storage disease is genetically caused and comparatively rare. However, the accumulated cases of all types is not so rare, but each one may be very difficult to diagnose. Some examples: Hurler’s disease 30 cases/year in the U. S. Sheie disease 10 cases/year in the U. S. Hurler/Sheie disease 30 cases/year in the U. S. There are about 28 variations of all of the lysosomal storage diseases.
HURLER’S DISEASE Let us take us one example, Hurler’s disease. This disease has its onset in infancy. There is a linear arrest in growth at ~1 year of age. There is psychomotor retardation. Since the GAGs pile up in the joints and internal organs, there is a distorted facial appearance, deformed and stiff joints with an enlarged liver and spleen. In the eyes, the cornea becomes cloudy and the optic nerve degenerates. Also glaucoma may occur. The disease is usually fatal within a few years due to congestive heart failure and respiratory pulmonary infections. Biochemically, this is the disease in which alpha-iduronidase is deficient. There is an accumulation of dermatan sulfate and heparan sulfate (2:1) in the tissues (including the eyes), blood and urine.
This diagram shows a typical sequence of degradation for the GAG: dermatan sulfate. Here you can see some of the enzymes associated with various GAG degradations. An important point is the early involvement of alpha-L-iduronidase in the degradative sequence (red arrow). This essentially leaves the molecule in a large, nearly intact form to pile up in tissues and fluids. The odd sequence of dermatan sulfate (in case you didn’t catch it [blue arrow]) is typical of GAGs.
Here are some typical appearances of Hurler disease children. The blank, unknowing stare – the swollen organs and joints are common. The cornea is seen at the right. In particular, note the ring of GAG deposits in the tissue.
This picture shows the accumulation of GAGs in a histiocyte of the brain of a Hurler’s patient. A histiocyte is a type of phagocyte derived from bone marrow that invades other tissues. Note the engorgement of GAGs in the vacuoles of the cell (one colored orange).
ASSAY FOR HURLER’S DISEASE In the assay, an artificial substrate replaces dermatan sulfate (or other GAG) with a fluorgen bound to iduronic acid. When the enzyme lyses the substrate, the product: 4-methylumbelliferone fluoresces at 446 nm in proportion to the activity of the enzyme.
TREATMENT • There are two forms of treatment: • bone marrow transplant. The earlier this is done the • better to avoid brain irreversible damage. The bone • marrow makes the missing enzyme and can be successful, but it is risky. • enzyme replacement therapy. This has limited • success and often will not correct neural damage. • It’s long term outcome is not known.
FOR REVIEW: • 1) Basic GAG structure and function . Do not memorize • specific structures. • 2) Can you explain the difference between a glycoprotein • and a proteoglycan? • 3) What is decorin and lumican? • 4) What is known about the nature of GAG/proteoglycans • and collagens in the spacing of collagen fibers in the • corneal stroma? How are collagens V/VI involved in • this association? What kind of a spacing structure • (geometric) is proposed for collagen and proteoglycan • spacing? • 5) What is known about hyaluronate and type II associations? • What about type IX/XI collagen there? • How does GAG degradation occur and where does it go • wrong in the mucopolysaccharidoses? • 7) Can you thoroughly explain Hurler’s disease? How is it assayed?