Download
1 / 24
240 likes | 1.02k Views
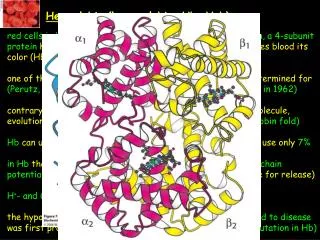
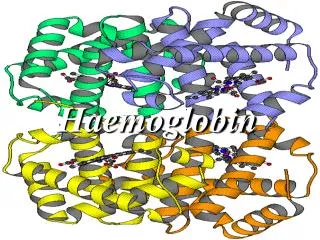
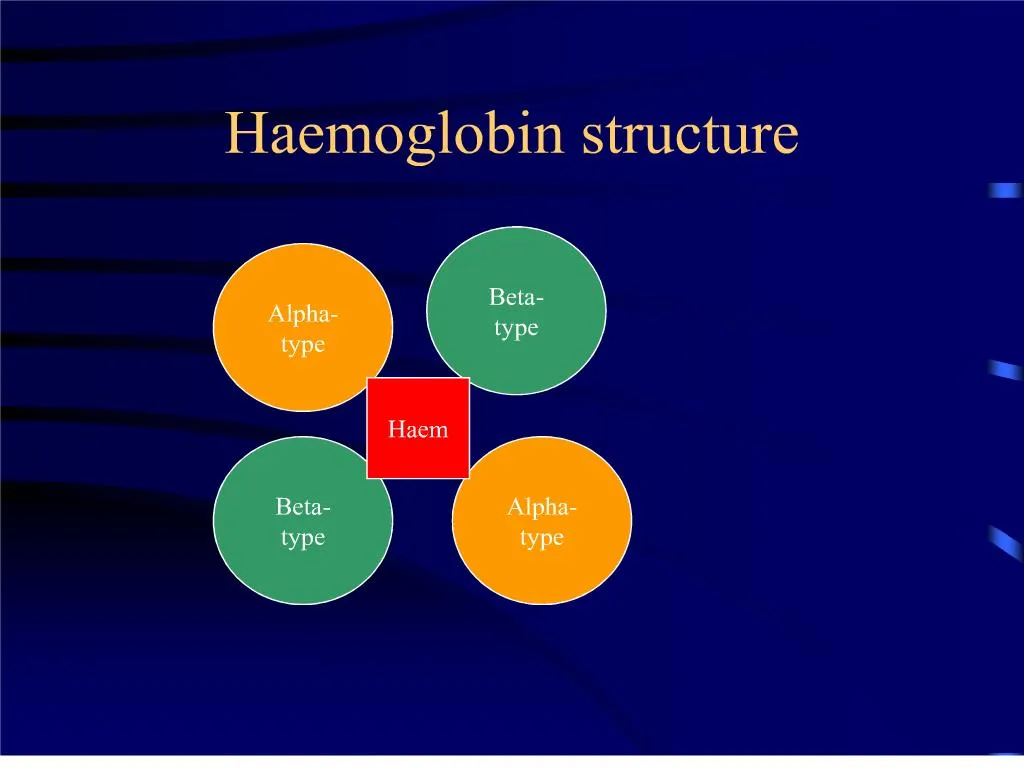
Normal Haemoglobin. Always 2 Beta-type and 2 Alpha-type globin chains carrying haem moleculeBeta-typeepsilon, gamma, beta, thetaAlpha-typezeta, alpha. Haemoglobin structure. So functional Hb is always a heterotetramerthere must be 2 Beta and 2 Alpha for oxygen carrying functiondifferent types at different stages of fetal and early neonatal lifeby 6 months we have adult proportions.

E N D
1. Haemoglobin structure
2. Normal Haemoglobin Always 2 Beta-type and 2 Alpha-type globin chains carrying haem molecule
Beta-type
epsilon, gamma, beta, theta
Alpha-type
zeta, alpha
4. Haemoglobin structure So functional Hb is always a heterotetramer
there must be 2 Beta and 2 Alpha for oxygen carrying function
different types at different stages of fetal and early neonatal life
by 6 months we have adult proportions
5. Hb development Up to 8/40
zeta2/epsilon2, alpha2/epsilon2, zeta2/gamma2
From 8/40 to birth
85% alpha2/gamma2 (HbF)
5-10% alpha2/beta2 (HbA)
remainder alpha2/theta2 (HbA2) + others
By 6/12, adult proportions of A, A2, F
6. Normal adult Hb HbA (alpha2/beta2)
97% +
HbA2 (alpha2/theta2)
2-3%
HbF (alpha2/gamma2)
0.5% or less
NOTE ALL NEED ALPHA!
7. Haemoglobin abnormalities Haemoglobinopathies
normal amounts of abnormal beta chains
crystalline disorders (S, C, D, E)
familial polycythaemia, M Hb, unstable Hb, HPFH
Thalassaemias
reduced amounts of normal alpha or beta chains
Can be BOTH!
8. Thalassaemias (simplistic) Reduced production of BETA chains
BETA thalassaemias
Reduced production of ALPHA chains
ALPHA thalassaemias
more severe clinical disease
9. Beta thalassaemias Beta chain deficiency
So reduced HbA
BUT retained production of other beta-type chains, so increased
theta production (HbA2)
gamma production (HbF)
10. Beta thalassaemias Encoded by a single gene pair
Autosomal recessive (but not totally)
heterozygotes have beta thalassaemia trait
homozygotes have beta thalassaemia (thalassaemia MAJOR)
but they are ALIVE at birth
variable clinical severity - why?
11. Inheritance of beta thalassaemia
12. Beta plus thalassaemia genes If the mutation causes total shutdown of the beta chain gene
no beta chain produced
Beta nought thalassaemia
If the mutation reduces beta chain production (but does not shut it down)
some beta chain produced
Beta plus thalassaemia
13. Combinations Beta/beta plus heterozygote
microcytosis, Hb normal
raised A2 and F
Beta/beta nought heterozygote
more severe microcytosis, Hb normal
raised A2 and F
14. Combinations Beta plus/beta plus
microcytosis, +/- anaemia
Beta nought/beta nought
microcytosis, red cell changes, transfusion dependent
Beta plus/beta nought
microcytosis, variably anaemic
15. Inheritance of alpha thalassaemia More complex as encoded by 2 gene pairs (so four genes per person, not two)
However, usually due to whole gene deletions, so total gene loss/shutdown
haematology and clinical presentation depends on how many genes are lost
16. Gene deletions in alpha thalassaemia
17. Gene deletions in alpha thalassaemia
18. Gene deletions in alpha thalassaemia
19. Clinical disorders Alpha/alpha +
alpha thalassaemia trait, normal Hb, normal or slightly reduced MCV
Alpha +/Alpha + or alpha/alpha 0
normal Hb, microcytic
20. Clinical disorders Alpha +/alpha 0
HbH disease, reduced Hb, splenomegaly, may or may not be transfusion dependent
presence of beta tetramers (HbH) on film (�golf ball� cells)
unlike in beta thalassaemia, there is no substitute for alpha
21. Hydrops fetalis Four gene deletion
no alpha chain production
incompatible with life
fetus dies in utero
gamma tetramers instead - Hb Barts
22. HbH and Hb Barts
23. Laboratory diagnosis Beta thalassaemia
relies on raised F and A2
Alpha thalassaemia
F and A2 normal
may see �golf balls� on HbH prep
gene analysis