Download
1 / 81
810 likes | 1.19k Views
第十一章 电解和库仑分析法 Electrolysis and Coulometry. 一 电解分析的基本原理. 电解分析法. 二 电解分析方法和应用. 一 库仑分析的基本原理. 库 仑 分 析. 二.控制电位库仑分析. 三 . 库仑滴定法. 电解分析法是将被测溶液置于电解装置中进行电解,使被测离子在电极上以金属或其它形式析出,由电解所增加的重量求算出其含量的方法。这种方法实质上是重量分析法,因而又称为电重量分析法。

E N D
第十一章 电解和库仑分析法Electrolysis and Coulometry
一 电解分析的基本原理 电解分析法 二 电解分析方法和应用 一 库仑分析的基本原理 库 仑 分 析 二.控制电位库仑分析 三. 库仑滴定法
电解分析法是将被测溶液置于电解装置中进行电解,使被测离子在电极上以金属或其它形式析出,由电解所增加的重量求算出其含量的方法。这种方法实质上是重量分析法,因而又称为电重量分析法。电解分析法是将被测溶液置于电解装置中进行电解,使被测离子在电极上以金属或其它形式析出,由电解所增加的重量求算出其含量的方法。这种方法实质上是重量分析法,因而又称为电重量分析法。 库仑分析法是在电解分析法的基础上发展起来的一种分析方法。它不是通过称量电解析出物的重量,而是通过测量被测物质在100%电流效率下电解所消耗的电量来进行定量分析的方法。
共同点: 分析时不需要基准物质和标准溶液,是一种绝对的分析方法,并且准确度高。 不同点: 电重量法只能用来测量高含量物质,而库仑分析法特别适用于微量、痕量成分的测定。
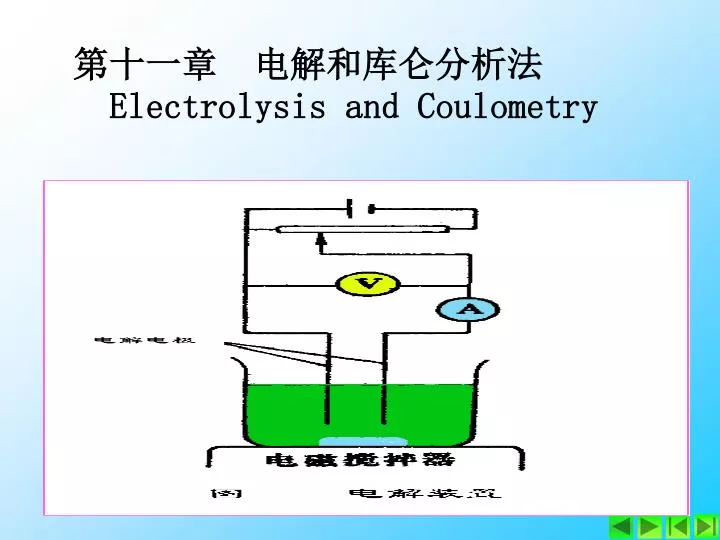
第一节 电解分析法 一 电解分析的基本原理 (一) 电解过程中电流和电压的关系 当外加电压很小时,有一个逐渐增加的微小电流通过电解池,这个微小电流称为残余电流(主要由电解液中杂质的电解产生)。当外加电压增大到某一数值时,电流迅速增大,并随着电压的增大直线上升,这时电解池内发生了明显的电极反应: 阴极反应 Cu2+ + 2e- Cu 阳极反应 2H2O O2 + 4H+ + 4e-
图11.1 分解电压 图11.2 电解装置
设在铂电极上电解硫酸铜溶液(见图)。当外加电压较小时,不能引起电极反应,几乎没有电流或只有很小电流通过电解池。如继续增大外加电压,电流略为增加,直到外加电压增加至某一数值后,通过电解池的电流明显变大。这时的电极电位称析出电位(φ析),电池上的电压称分解电压(E分)。而发生的电解现象是,阴极上Cu2+离子比H+离子更易被还原设在铂电极上电解硫酸铜溶液(见图)。当外加电压较小时,不能引起电极反应,几乎没有电流或只有很小电流通过电解池。如继续增大外加电压,电流略为增加,直到外加电压增加至某一数值后,通过电解池的电流明显变大。这时的电极电位称析出电位(φ析),电池上的电压称分解电压(E分)。而发生的电解现象是,阴极上Cu2+离子比H+离子更易被还原 Cu2++2e = Cu 阳极上 H2O中 OH-离子被氧化 2H2O - 4e →O2↑+ 4H+ (二) 分解电压和析出电位
如将电源切断,这时外加电压虽已经除去,但伏特计上的指针并不回到零,而向相反的方向偏转,这表示在两电极间仍保持一定的电位差。这是由于在电解作用发生时,阴极上镀上了金属铜,另一电极则逸出氧。金属铜和溶液中的Cu2+组成一电对,另一电极则为氧的电极。当这两电对连接时,形成一原电池。此原电池的反应方向是由两电对的电极电位的大小决定的。原电池发生的反应为如将电源切断,这时外加电压虽已经除去,但伏特计上的指针并不回到零,而向相反的方向偏转,这表示在两电极间仍保持一定的电位差。这是由于在电解作用发生时,阴极上镀上了金属铜,另一电极则逸出氧。金属铜和溶液中的Cu2+组成一电对,另一电极则为氧的电极。当这两电对连接时,形成一原电池。此原电池的反应方向是由两电对的电极电位的大小决定的。原电池发生的反应为 负极 Cu-2e→Cu2+ 正极 O2+4H++4e→2H2O 反应方向刚好与电解反应相反。 设溶液中CuSO4和H+离子浓度均为lmol/L,此原电池的电动势为 ε=φ正-φ负=1.23-0.35=0.89V
可见,电解时产生了一个极性与电解池相反的原电池,其电动势称为“反电动势”(ε反)。因比,要使电解顺利进行,首先要克服这个反电动势。可见,电解时产生了一个极性与电解池相反的原电池,其电动势称为“反电动势”(ε反)。因比,要使电解顺利进行,首先要克服这个反电动势。 理论分解电压E分为 E分=ε反=φ正-φ负 对于电解1mol/LCuSO4溶液,其E分不为0.89V, 而为1.49V。 图11.3 金属电沉积装置
原因:一是由于电解质溶液有一定的电阻,欲使电流通过,必须用一部分电压克服iR(i为电解电流,R为电解回路总电阻)电位降,一般这是很小的;二是主要用于克服极化现象产生的阳极反应和阴极反应的过电位(η阳和η阴)。 因此,电解lmol/LCuSO4溶液时,需要外加电压E分=1.49V,而不是E分=0.89V。多加的0.60V,就是用于克服iR电位降和由于极化产生的阳极反应和阴极反应的过电位。 让我们来讨论这个(0.60V)过电压和过电位:
E外=E分+iR =[(φ平(阳)+ηa)-(φ平(阴)+ηc)]+iR 通常,可设iR→0,则 E外=E分=(φ平(阳)+ηa)-(φ平(阴)+ηc) 定量关系
有两种极化作用产生的过电位 过电位可分为浓差过电位和电化学过电位两类。前者是由浓差极化产生的,后者是由电化学极化产生的。电化学极化是由电化学反应本身的迟缓性所引起的。一个电化学过程实际上由许多分步过程所组成,其中最慢一步对整个电极过程的速度起决定性的作用。在许多情况下,电极反应这一步的速度很慢,需要较大的活化能。因此,电解时为使反应能顺利进行,对阴极反应而言,必须使阴极电位比其平衡电位更负一些;对阳极反应而言,则必须使阳极电位比其平衡电位更正一些。这种由于电极反应引起的电极电位偏离平衡电位的现象,称为电化学极化。电化学极化伴随产生过电位。可分析如下:
由于电池回路的电压降和阴、阳极的极化所产生的超电位,使得实际上的分解电压要比理论分解电压大。使电解反应按一定速度进行所需的实际电压称为实际分解电压,U=(c-a)+(c-a)+iR,为使电极反应向非自发方向进行,外加电压应足够大,以克服电池反电动势,是实际分解电压。由于电池回路的电压降和阴、阳极的极化所产生的超电位,使得实际上的分解电压要比理论分解电压大。使电解反应按一定速度进行所需的实际电压称为实际分解电压,U=(c-a)+(c-a)+iR,为使电极反应向非自发方向进行,外加电压应足够大,以克服电池反电动势,是实际分解电压。 理论分解电压: 通常将两电极上产生迅速的连续不断的电极反应所需的最小电压称为理论分解电压,因此理论分解电压即电池的反电动势,U=c-a 。
二 电解分析方法和应用 (一)、控制电流电解法(Constant current electrolysis) 1.过程:控制电解电流保持不变,随着电解的进行,外加电压不断增加,因此电解速度很快。 特点:电解速度快,但选择性差。 去极剂:加入阴极或阳极去极剂可以克服选择性差的问题。如在电解Cu2+时,为防止 Pb2+同时析出,可加入NO3- 作阴极去极剂。此时 NO3- 可先于Pb2+析出。
基本装置 图11.4 控制电流电解装置
2.阴极电位随时间变化曲线 随着电解的进行,阴极表面附近Mn+浓度不断降低,为了维持电解电流恒定,就必须增大外加电压,使阴极电位更负。这样由于静电引力作用使Mn+以足够快的速度迁移到阴极表面,并继续发生电极反应以维持电解电流恒定, Mn+继续在阴极上还原析出,直到电解完全。 Ec 图11.6 控制电流电解曲线
3.特点及其应用范围 电解效率高,分析速度快,选择性差。只能用于溶液中有一种可还原金属离子的定量分析, 用于分离金属活动顺序氢两侧的金属元素。
(二)、控制电位电解分析 1. 控制外加电压 例如:0.1M H2SO4介质中,含1.0M Cu2+ 和 0.01M Ag+,问能否通过控制外加电压方法使二者分别电解而不相互干扰? a) 各离子在阴极的析出电位 Cu的析出电位: Cu = Cu +0.059/2 lg cCu2+ = 0.337V Ag的析出电位: Ag=Ag+0.059lg cAg+ =0.779+0.059lg0.01=0.681V 因为Ag > cu ,故Ag2+先于Cu2+析出。
b) Ag完全析出时的外加电压 设Ag2+“完全”析出时溶液中Ag2+的浓度为10-6M,则此时Ag的阴极电位:Ag= 0.779+0.059lg10-6 = 0.445V O2的阳极电位: O2=+0.059/2lg(PO21/2cH+)+=1.23+0.059/2lg(11/20.22)+0.72=1.909V因此,Ag完全析出时的外加电压=1.909 - 0.445V = 1.464V c)Cu开始析出时的外加电压=1.909-0.337=1.572V 可见在Ag完全析出时的电压并未达到Cu析出时的分析电压。即此时Cu不析出或者说Cu不干扰测定。即可以通过控制外加电压来进行电解分析。
图11.7 自动控制电极电位装置 A:辅助电压;Amp:放大;M:可逆电机
2、控制阴极电位电解分析 当试样中存在两种以上离子时,随着电解反应的进行,离子浓度将逐渐下降,电池电流也逐渐减小,此时通过外加电压方式达不到好的分别电解的效果。即第二种离子亦可能被还原,从而干扰测定。因此,常以控制阴极电位的方式进行电解分析。 将工作电极(阴极)和参比电极放入电解池中,控制工作电极电位(或控制工作电极与参比电极间的电压)不变。开始时,电解速度快,随着电解的进行,浓度变小,电极反应速率,当i=0时,电解完成。
3.电流—时间曲线 电流随时间增长而不断减小,由于残余电流的存在,电流最后达到恒定的背景电流值。 电流效率为100%时,电流和时间的关系为: it=i0· 10-kt 浓度和时间的关系: ct=c0· 10-kt k为常数,它与电极表面积、溶液体积、搅拌速度以及电极反应类别等因素有关。 i/A t/min 图11.8 控制阴极电位电解的i-t曲线
4. 特点及应用 由于控制阴极电位能有效地防止共存离子的干扰,因此选择性好。该法既可作定量测定,又可以广泛地用作分离技术,常用于多种金属离子共存情况下某一离子含量的测定。
(三) 汞阴极电解法 前述电解分析的阴极都是以Pt作阴极,如果以 Hg作阴极即构成所谓的Hg阴极电解法。但因 Hg密度大,用量多,不易称量、干燥和洗涤,因此只用于电解分离,而不用于电解分析。 特点: 1)可以与沉积在 Hg上的金属形成汞齐; 2)H2 在 Hg上的超电位较大,扩大电解分析电压范围;3)Hg比重大,易挥发除去。 这些特点使该法特别适合用于分离。
应用例子: 1)Cu、Pb、Cd在 Hg阴极上沉积而与U 分离; 2)伏安分析和酶法分析中高纯度电解质的制备;3)消除钢铁中大量铁,以利于其中微量样品的测定等。
库仑分析是基于电量的测量,因此,通过电解池的电流必须全部用于电解被测的物质,不应当发生副反应和漏电现象,即保证电流效率100%,这是库仑分析的关键。如一个测定只包含初级反应,即被测物质是直接在电极上发生反应的,称为初级库仑分析。在初级库仑分析中,只要求电化学反应定量进行。如测定要靠次级反应来完成,即被测物质是间接与电极电解产物进行定量反应的,称为次级库仑分析。这时,不但要求电极反应定量发生,而且要保证次级反应定量进行。常用的库仑分析与电重量分析一样,分为控制电位库仑分析和控制电流库仑分析两类,后者又称为库仑滴定。库仑分析是基于电量的测量,因此,通过电解池的电流必须全部用于电解被测的物质,不应当发生副反应和漏电现象,即保证电流效率100%,这是库仑分析的关键。如一个测定只包含初级反应,即被测物质是直接在电极上发生反应的,称为初级库仑分析。在初级库仑分析中,只要求电化学反应定量进行。如测定要靠次级反应来完成,即被测物质是间接与电极电解产物进行定量反应的,称为次级库仑分析。这时,不但要求电极反应定量发生,而且要保证次级反应定量进行。常用的库仑分析与电重量分析一样,分为控制电位库仑分析和控制电流库仑分析两类,后者又称为库仑滴定。 第二节 库仑分析
(一) Faraday定律 电解过程中,在电极上析出的物质的重量与通过电解池的电量之间的关系,遵守Faraday定律,可用下式表示 一 库仑分析的基本原理 式中:W为物质在电极上析出的克数,M为分子量,n为电子转移数,F为Faraday常数,1F=96487C,Q为电量,以C为单位。如通过电解池的电流是恒定的,则 Q=It将上式代入,得:
Faraday定律的正确性已被许多实验所证明。它不仅可应用于溶液和熔融电解质,也可应用于固体电解质导体。Faraday定律的正确性已被许多实验所证明。它不仅可应用于溶液和熔融电解质,也可应用于固体电解质导体。 如电流不恒定,而随时间不断变化,则
在一定的外加电压条件下,通过电解池的总电流 iT,实际上是所有在电极上进行反应的电流的总和。它包括: (1) 被测物质电极反应所产生的电解电流ie; (2) 溶剂及其离子电解所产生的电流is; (3) 溶液中参加电极反应的杂质所产生的电流iimp。 电流效率ηe为 ηe=ie/(ie+is+iimp)×100%=ie/ iT ×100% (二).电流效率
(三)影响电流效率的主要因素 1.溶剂的电极反应 2.电活性杂质在电极上的反应 3.溶液中可溶性气体的电极反应 4.电极自身参与反应 5.电解产物的再反应 6.共存元素电解 电流效率为:
(一)基本原理 在电解池装置的电解电路中串入一个能精确测量电量的库仑计。电解时,用恒电位装置控制阴极电位,以100%的电流效率进行电解,当电流趋于零时,电解即完成。由库仑计测得电量,根据Faraday定律求出被测物质的含量。 图11.9 库仑分析的电位仪 二.控制电位库仑分析 Controlled potential Coulometry
图11.10自动控制电极电位装置 A:辅助电压;Amp:放大;M:可逆电极
氢氧库仑计和银库仑计等是一种最基本、最简单而且最准确的库仑计。氢氧库仑计是一个电解水的装置,电解管与刻度管用橡皮管连接。电解管中焊两片铂电极,管外为恒温水浴套。电解液可用0.5mol/LK2SO4或Na2SO4,通过电流时在阳极上析出氧气氢氧库仑计和银库仑计等是一种最基本、最简单而且最准确的库仑计。氢氧库仑计是一个电解水的装置,电解管与刻度管用橡皮管连接。电解管中焊两片铂电极,管外为恒温水浴套。电解液可用0.5mol/LK2SO4或Na2SO4,通过电流时在阳极上析出氧气 H2O-2e→ 1/2O2 +2H+ 在阴极上析出氢气 2H+2e→H2 总反应为 H2O → H2↑+1/2O2↑ 1.气体库仑计
在标准状况下,每库仑电量析出O.1741mL氢、氧混合气体(实际运算用O.1739mL)。这种库仑计使用简便,能测量1O C以上的电量,准确度达O.1%以上,但灵敏度较差。
2.积分法和电子积分库仑计 现代仪器多采用积分运算放大器库仑计或数字库仑计测定电量。在电解过程中可记录Q(t)-t曲线,由
算出Q。 即算出了所通过的电量。 用做图法求i0与k的关系,即 lgit=lgio+(-kt)
控制电位库仑分析法具有准确、灵敏、选择性高等优点,特别适用于混合物质的测定,因而得到了广泛的应用。可用于五十多种元素及其化合物的测定。其中包括氢、氧、卤素等非金属,钠、钙、镁、铜、银、金、铂族等金属以及稀土和镧系元素等。控制电位库仑分析法具有准确、灵敏、选择性高等优点,特别适用于混合物质的测定,因而得到了广泛的应用。可用于五十多种元素及其化合物的测定。其中包括氢、氧、卤素等非金属,钠、钙、镁、铜、银、金、铂族等金属以及稀土和镧系元素等。 在有机和生化物质的合成和分析方面的应用也很广泛,涉及的有机化合物达五十多种。例如,三氯乙酸的测定,血清中尿酸的测定,以及在多肽合成和加氢二聚作用等的应用。 控制电位库仑法也是研究电极过程、反应机理等方面的有效方法。测定电极反应的电子数不需事先知道电极面积和扩散系数。 (二)特点及应用
例如,在100mL 0.lmol/L HCl中,以银为阳极,汞滴为阴极,-O.65V(vs. SCE)时电解O.0399mmol/L苦味酸,利用氢氧库仑计测得电量为65.7C,求出电极反应电子数n=17.07,证明了苦味酸的还原反应为
电解实验条件 1)电流密度:电流密度过小,析出物紧密,但电解时间长;电流密度过大,浓差极化大,可能析出H2,析出物疏松----通常采用大面积的电极(如网状Pt电极); 2)搅拌及加热;
3)pH和配合剂:PH过高,金属水解,可能析出待测物的氧化物;PH过低,可能有H2析出。当在碱性条件下电解时,可加入配合剂,待测离子保留在溶液中。如电解沉积Ni2+,在酸性或中性都不能使其定量析出,但加入氨水后,可防止H2析出,形成Ni(NH3)42+可防止Ni(OH)2沉淀。3)pH和配合剂:PH过高,金属水解,可能析出待测物的氧化物;PH过低,可能有H2析出。当在碱性条件下电解时,可加入配合剂,待测离子保留在溶液中。如电解沉积Ni2+,在酸性或中性都不能使其定量析出,但加入氨水后,可防止H2析出,形成Ni(NH3)42+可防止Ni(OH)2沉淀。 4)去极剂:加入比干扰物更易还原的高浓度离子,可防止干扰离子析出。
由恒电流发生器产生的恒电流通过电解池,被测物质直接在电极上反应或在电极附近由于电极反应产生一种能与被测物质起作用的试剂,当被测物质作用完毕后,由指示终点的仪器发出信号,立即关掉计时器。由电解进行的时间t(S)和电流强度(A),可求算出被测物质的量W(g)。此法又称为控制电流库仑滴定法,简称为库仑滴定法。由恒电流发生器产生的恒电流通过电解池,被测物质直接在电极上反应或在电极附近由于电极反应产生一种能与被测物质起作用的试剂,当被测物质作用完毕后,由指示终点的仪器发出信号,立即关掉计时器。由电解进行的时间t(S)和电流强度(A),可求算出被测物质的量W(g)。此法又称为控制电流库仑滴定法,简称为库仑滴定法。 三. 库仑滴定法 Coulometric Titration (一)库仑滴定法基本原理
这种方法并不测量体积而测量电量。它与普通容量分析法突出的不同点在于,滴定剂不是由滴定管向被测溶液中滴加,而是通过恒电流电解在试液内部产生,产生滴定剂的量又与电解所消耗的电量成正比。因此,可以说库仑滴定是一种以电子作一滴定剂的容量分析。这种方法并不测量体积而测量电量。它与普通容量分析法突出的不同点在于,滴定剂不是由滴定管向被测溶液中滴加,而是通过恒电流电解在试液内部产生,产生滴定剂的量又与电解所消耗的电量成正比。因此,可以说库仑滴定是一种以电子作一滴定剂的容量分析。 例如:在酸性介质中测定Fe2+的含量,其i-E曲线,如图所示。工作电极可用铂电极,电流控制在1~100mA(通常为10mA)。开始阳极反应为 Fe2++= Fe3+ + e-
图11.13在酸性介质中Fe2+被氧化的i-E曲线 1.Fe2+ 2.Fe2++过量的Ce3+
由于反应的进行,电极表面上Fe3+离子浓度不断增加,Fe2+离子浓度不断下降,因而阳极电位将逐渐向正方向移动。最后,溶液中Fe2+还没有全部氧化为Fe3+,而阳极电位已达到了水的分解电位,这时在阳极上同时发生下列反应而析出氧由于反应的进行,电极表面上Fe3+离子浓度不断增加,Fe2+离子浓度不断下降,因而阳极电位将逐渐向正方向移动。最后,溶液中Fe2+还没有全部氧化为Fe3+,而阳极电位已达到了水的分解电位,这时在阳极上同时发生下列反应而析出氧 2H2O= O2↑+ 4H+ +4e 显然,由于上述反应的发生,使Fe2+离子氧化反应的电流效率低于100%,因而使测定失败。 如在溶液中加入过量的辅助电解质Ce3+离子,则Fe2+离子可在恒电流下电解完全。开始阳极上的主要反应为Fe2+氧化为Fe3+。
当阳极电位正移至一定数值时,Ce3+离子开始被氧化为Ce4+离子,而所产生的Ce4+,则转移至溶液主体,并氧化溶液中的 Fe2+离子。由于Ce3+过量,稳定了电极电位,防止了水的电解。根据反应可知,阳极上虽发生了Ce3+的氧化反应,但其所产生的Ce4+又将Fe2+氧化为Fe3+。因此,电解所消耗的总电量与单纯Fe2+完全氧化为Fe3+的电量是相当的。可见,用这种间接库仑分析方法,既可将工作电极的电位稳定,防止发生副反应,又可使用较大的电流密度,以缩短滴定的时间。
图11.14 库仑滴定的装置 T:电钟; R1:标准电阻; M:电池搅拌器; V:恒电流电源;R2:高电阻;ROT:电位计; G:检流计; EI:终点指示;K:开关; e:指示电极 (二)库仑滴定的装置
1.恒电流电源 2.计时装置 3.库仑计 4.指示装置