Download

1 / 26
290 likes | 801 Views
Motor Neurone Disease. Sarah Bray 23 rd April 2013. Aims. Background Presentation Signs Diagnostic criteria Differential Diagnoses Investigation Research Symptoms Symptomatic Management Preparation and support Challenges for the GP. Curriculum Statement.

E N D
Motor Neurone Disease Sarah Bray 23rd April 2013
Aims • Background • Presentation • Signs • Diagnostic criteria • Differential Diagnoses • Investigation • Research • Symptoms • Symptomatic Management • Preparation and support • Challenges for the GP
Curriculum Statement 3.18 Care of People with Neurological Problems • 1.2 Know the epidemiology of common and/or important neurological conditions such as epilepsy, headache and facial pain syndromes, brain infections, neurological causes of vertigo, spinal cord disease, spinal root compression/irritation, peripheral neuropathies, multiple sclerosis, motor neurone disease, Parkinson’s disease and common and/or important movement disorders, brain tumours, and common and/or important inherited and congenital conditions
Mrs K • 76 year old lady admitted to MAU with increasing breathlessness, dysphasia, dysphagia and ataxic gait • Previous diagnosis of lacunar stroke but symptoms progressing • On examination thenar and hypothenar wasting, clawing of fingers, fasciculation of arms, hands and tongue and weak, nasal speech • Suspected respiratory muscle weakness and aspiration pneumonia • Seen by neurology – diagnosed with suspected MND, awaiting nerve conduction studies
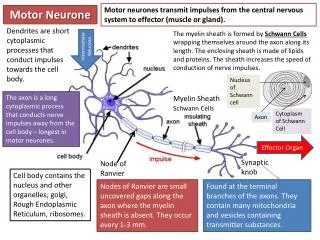
Background • Most cases due to amyotrophic lateral sclerosis (ALS) • Also progressive bulbar palsy, progressive muscular atrophy and primary lateral sclerosis • Family of diseases of unknown aetiology • Progressive degeneration of upper and/or lower motor neurones • Leading to wasting of muscles and weakness • Average survival 40% at 5 years, medial survival 3-5 years • Mean age of onset 56 years • Male : female ratio = 2 : 1 • Annual incidence 2 per 100,000 • Prevalence 5-7 per 100,000
Presentation • ALS • Mixed upper and lower motor neurone involvement • 3 recognised patterns: • Limb onset (common) • Bulbar onset (20%) • Respiratory onset (least common) • Primary lateral sclerosis • Pure UMN features • Progressive muscular atrophy • Pure LMN features
Signs • Upper motor neurone • Generalised spasticity, hyperreflexia, pathologic reflexes, up-going plantar responses and often emotional lability (i.e. pseudobulbar affect) • Lower motor neurone • Weakness, muscle wasting, reduced muscle tone, hyporeflexia and fasciculation • Involvement of bulbar innervated nerves • Dysarthria and dysphagia
Diagnostic criteria • Presence of: • Evidence of LMN degeneration by clinical, electrophysiological or neuropathological examination. • Evidence of UMN degeneration by clinical examination. • Progressive spread of symptoms or signs within a region or to other regions, as determined by history or examination. • Together with the absence of: • Electrophysiological and pathological evidence of other disease processes that might explain the signs of LMN and/or UMN degeneration. • Neuroimaging evidence of other disease processes that might explain the observed clinical and electrophysiological signs.
Differential Diagnosis • Important diseases that can mimic MND: • Benign cramp fasciculation syndrome • Cervical radiculomyelopathy (multilevel degenerative disease of the cervical spine) • Dual pathologies • Multifocal motor neuropathy with conduction block • Inclusion body myositis
… conditions sharing similar features • Diabetic amyotrophy. • Guillain-Barré syndrome. • Post-polio syndrome. • Spinal cord tumours. • Glioma of brainstem. • Viral plexopathies. • Lyme disease. • Post-radiation myeloplexopathy. • Tay-Sachs disease (adult form). • Cerebrovascular disease and stroke. • Polymyositisor dermatomyositis. • HIV-associated neuropathy/myopathy/radiculopathy (as one of the complications of HIV). • Lepto-meningeal disease, eg due to carcinomatosis or vascular collagen disease. • Spinal muscular atrophy (Kennedy's syndrome). • Hereditary polyneuropathies, e.g. Charcot-Marie-Tooth syndrome. • Focal muscular atrophies (monomelicamyotrophy). • Myasthenia gravis or Lambert-Eaton myasthenic syndrome. • Peripheral nerve lesions, particularly due to diabetic neuropathy. • Thyrotoxicosis with associated myopathy.
Investigations • Electrophysiological studies • Electromyography and nerve conduction studies • Exclude other pathologies • CT/MRI brain and/or spine • Bloods • Muscle biopsy
Research • Stem-cell transplantation • Autologous mesenchymal stem cells • Allogenic haemopoietic stem cells • Antioxidants
Medical Management • Riluzole • Neuroprotective glutamate-release inhibitor • Only drug approved for treatment (NICE) • Slows disease progression • Extends survival and tracheostomy-free life by an average of 3 months • Requires 3 monthly LFTs • MDT input • Neurologist, physiotherapy, OT, SALT, dietician, case manager, GP, district nurses, social worker, palliative care team
Symptoms • Weakness (100%) • Constipation (65%) • Pain (50-60%) • Cough (50-60%) • Insomnia (40-50%) • Breathlessness (40-50%) • Dribbling (30-40%) • Pseudobulbar affect (up to 50%) • Anxiety and depression • Mobility problems • Communication difficulties • Psychosocial issues
Symptom management • Weakness • Try to maintain comfort, prevent contractures and joint dislocation • Physiotherapy • Insomnia • Try to isolate the cause • Cautious use of night sedation • Respiratory insufficiency/breathlessness • NIV (NIPPV, BiPAP) • Treat infections • Cough-assist device/physiotherapy
Symptom Management • Pseudobulbar affect • SSRI, amitriptyline • Pain • Management of cause: • Stiff joints – careful positioning/physiotherapy • Inflammation – NSAID • Joint pains – intra-articular steroid injections • Muscle cramp – quinine sulphate (tonic water) • Muscle spasm – diazepam/baclofen • Skin pressure – regular turning, analgesic ladder • Sensory disturbance - amitriptyline
Symptom Management • Dysphagia • SALT and dietetic input • Ice packs or chips • Artificial feeding (NG/RIG/PEG) • Dysarthria • SALT –visual aids, lightwriter, other communication aids • Choking • SALT - swallowing techniques • Suction • Main fear of patients
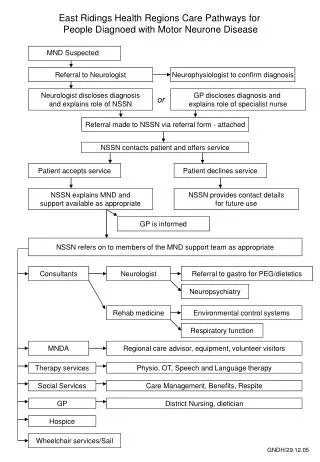
Preparation and Support • Discussion in advance about the patients wishes regarding artificial nutrition and respiratory support, and in what circumstances to stop these • Breathing Space Kit from MNDA • Anticipatory meds • Support for patient and carer(s) • Palliative care services • Day therapy/respite
Mrs K • PEG inserted in the month following diagnosis • Attended multidisciplinary MND clinic • Struggling with secretions – hyoscine hydrobrominde and amitriptyline tried, botox injections to parotid glands suggested • NIV supplied but Mrs K reluctant to use
Mrs K • 4 months later admitted to hospice with sudden deterioration in mobility • Recent antibiotic course for UTI • Carers QDS but husband struggling • Speech now very poor • Requires hoisting for all transfers • Very tearful
Mrs K • Difficult to understand speech but one thing clear, patient consistently requests to go home • Very emotional and tearful • Husband purposefully did not visit for the first 5 days as he felt he needed a break • Via telephone contact he felt that if she was becoming increasingly dependent he could not manage at home and she needed to go into 24 hour care • Deprivation of liberty vs maintaining safety of patient
Mrs K • Efforts made to maximise communication – lightwriter • Independent advocates provided by family support team for both patient and husband • Antibiotics switched but Mrs K continued to spike temperatures • Discussion with family that Mrs K was less well • Husband and son came to visit at the hospice, decision made not for IV antibiotics or transfer to hospital • Mrs K passed away peacefully in the presence of her husband and daughter
Challenges for the GP • Insidious onset • Lack of information • No cure and minimal effective treatment • Progressive nature of the condition • Ethical issues surrounding artificial nutrition and assisted ventilation • Communication difficulties • Effective palliation • Support for carers
Patient Perspective When I wake each morning I decide … This can be a good day or a bad day – my choice. I can be happy or sad – my choice. I can complain or I can cope – my choice. Life can be a chore or a challenge – my choice. I can take from life or give to life – my choice. If all things are possible, How I deal with those possibilities is – my choice. Steve Shackel, diagnosed with MND