Download

1 / 47
470 likes | 598 Views
Introduction to Sequence Analysis. MARC: Developing Bioinformatics Programs July, 2008 Alex Ropelewski ropelews@psc.edu Hugh Nicholas nicholas@psc.edu. Bioinformatics.

E N D
Introduction to Sequence Analysis MARC: Developing Bioinformatics Programs July, 2008 Alex Ropelewski ropelews@psc.edu Hugh Nicholas nicholas@psc.edu These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Bioinformatics The interdisciplinary science of using computational approaches to analyze, classify, collect, represent and store biological data with the goal of accelerating and enhancing the understanding of DNA, RNA and Protein sequences.
Sequence Analysis Process of applying computational methods to a biological molecule represented as a character string. The goal is to infer information about the structure, function, or evolutionary history of the sequence.
What is a Sequence? • A sequence is a way to represent a protein, DNA, or RNA molecule as a character string. Phospholipase A2 - Bos taurus (Bovine). MRLLVLAALLTVGAGQAGLNSRALWQFNGMIKCKIPSSEPLLDFNNYGCYCGLGGSGTPV DDLDRCCQTHDNCYKQAKKLDSCKVLVDNPYTNNYSYSCSNNEITCSSENNACEAFICNC DRNAAICFSKVPYNKEHKNLDKKNC
Why study families of sequences? • Families share a common function, structure, and are related through evolution Aldehyde Dehydrogenase Family Members
The Goal CURATED FAMILY: • All related sequences sharing a common function (Homologous Sequences) • All substantial motifs • Evolutionary history • Structural information • Experimental information These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns The Process Homology Modeling CURATED DATASET These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
The Toolkit GenBank Blast Clustalw Meme EMBL Fasta T-Coffee Mast UniProt Smith-Waterman MSA hmmer Pfam Needleman-Wunsch Probcons Profile-ss PDB Figtree Phylip Notung PDB Python BioPython Genedoc
The Toolkit Which is the proper tool for the task?
The Project • Part I: Submit three candidate families for your course project. • Part II: Collect an initial set of sequences, generate a multiple sequence alignment • Part III: Improve the quality of your alignment, and identify additional family members • Part IV: Add structural and/or evolutionary information, and give a final report These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns Part I Homology Modeling CURATED DATASET These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Evolutionary Analysis Hidden Markov Model Multiple Sequence Alignment Initial Query Profile & PSSM Local Patterns Part II Structural Libraries Homology Modeling CURATED DATASET Classification Libraries Sequence Libraries These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Initial Query Part III Homology Modeling CURATED DATASET Multiple Sequence Alignment Sequence Libraries Local Patterns Profile & PSSM These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Hidden Markov Model Classification Libraries Initial Query Profile & PSSM Sequence Libraries Local Patterns Part IV Structural Libraries Evolutionary Analysis Homology Modeling CURATED DATASET Multiple Sequence Alignment These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
What is an Information Library? • A compilation of prior experimental knowledge about biologically relevant molecules into a computer system. • Bioinformatics power is in the ability to leverage and apply this prior experimental knowledge to additional biological problems. • From a biologists prospective, there are different ways that we can organize this prior experimental knowledge: • Sequence • Structure • Family/Domain • Species • Taxonomy • Function/Pathway • Disease/Variation • Publication Journal • And many other ways
Structural Libraries • Structure libraries contain the actual three dimensional coordinates of a macromolecule. • Used to: • Determine if the three dimensional structure for a molecule has been solved • Visualize the three dimensional structure • Assist in homology modeling These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Classification (Family/Domain) Libraries • Built from sets of related sequences and contain information about the residues that are essential to the structure/function of the group of related sequences • Used to: • Generate a testable hypothesis that the query sequence belongs to the group. • Quickly identify a good group of sequences known to share a biological relationship. These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Classification Libraries - Representation • Consensus • Residue most common at each position in alignment • Composite • Set representation: (e.g. a {gc} x {acg} t a) • Composition Matrix • Table of how many residues present at each position • Position Specific Scoring Matrix (PSSM or Profile) • Log-odds likelihood of each residue at each position • Hidden Markov Model • Probabilistic state representation These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Sequence Libraries • Compilations of known sequences with experimental information about those sequences. • Used to: • Generate a testable hypothesis that the query sequence may be related to known sequences in the library. • Retrieve annotation information about sequences These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Searching Sequence Libraries • What question are you trying to answer? (Part 1) • Local: A part of one sequence is similar to a part of another sequence • Methods: Blast, Fasta, Smith-Waterman, Waterman-Egert “Maxsegs" • Global: The sequences are similar across their entire lengths • Methods: Needleman-Wunsch, Sellers These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
What Question Are You Trying To Answer? These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Searching Sequence Libraries • What question are you trying to answer? (Part 2) • You have a single sequence and want to find sequences related to that sequence • Search database with the sequence • You have a set of related sequences and want to find additional, distantly related sequences. • Search database with an abstract representation of the set of sequences These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Sequence Libraries – Results Searching with a Single Sequence These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Sequence Libraries – ResultsSearching with an Abstract Representation These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Multiple Sequence Alignment • An MSA is an alignment of a group of related sequences across their entire lengths in a manner than highlights the conservation of the important residues in the sequences • Critical building block for many next steps such as finding distantly related sequences, determining the evolutionary history, and homology modeling • Not all sets of related sequences can be aligned across their entire lengths cleanly These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Multiple Sequence Alignment These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Abstracting Multiple Sequence Alignments • Help locate distantly related sequences • Help resolve sequences that are not considered “statistically significant” by a database search, but may be homologous with the query sequence • Two common abstraction techniques: • PSSM – Position Specific Scoring Matrix (or Profile) • HMM – Hidden Markov Model
Position Specific Scoring Matrix (PSSM) • Think of a PSSM as a custom PAM or BLOSUM scoring matrix that has been specially tuned to locate sequences exactly like those in the alignment. • Log-odds likelihood of each residue at each position • Good MSA = Good PSSM • Poor MSA = Poor PSSM • A lot of Sequences = Good PSSM • Few Sequences = Good PSSM These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Hidden Markov Model (HMM) • Think of a HMM as a way to represent a multiple sequence alignment by deriving probabilities directly from the multiple sequence alignment • Probabilistic model – Includes probabilities for insertions and deletions • Good MSA = Good HMM • Poor MSA = Poor HMM • A lot of Sequences = Good HMM • Few Sequences = Poor HMM These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Improving a Multiple Sequence Alignment • When aligning groups of related sequences it is important to note that residues in those sequences are either: • Conserved (not mutated) • Unconstrained (when mutated can be almost any amino acid) • Constrained (when mutated must be one of a few amino acids) • Motifs are distinct units that consists of theconserved and constrained regions; • Motifs generally tell us the residues that are essential to the structure/function of the sequence • Typically, multiple sequence alignments contain motifs as well as unconstrained regions. • Aligning motifs in a multiple sequence alignment will improve the quality of the alignment These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Finding Motifs • Best method combines two theoretical methods derived from statistics: • EM (Expectation-Maximization) • We can recognize a good pattern, but don’t know where the patterns or motifs are within the set of sequences • Stochastic Sampling to reduce the volume of alignment space that must be searched • Too many possible motifs to exhaustively search • Exploit the “memory” of empirical position specific scoring matrix representation • A sequence segment that is part of the pattern will generally have a higher than “average” or expected score when scored using the matrix
Homology Modeling • Predicts the three-dimensional structure of a given protein sequence (TARGET) based on an alignment to one or more known protein structures (TEMPLATES) • If similarity between the TARGET sequence and the TEMPLATE sequence is detected, structural similarity can be assumed. • Homology Modeling can also be used to fill in missing information from an incomplete three-dimensional structure. These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Homology Modeling – Why it works Structural Superposition of Aldehyde Dehydrogenase Family Members These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
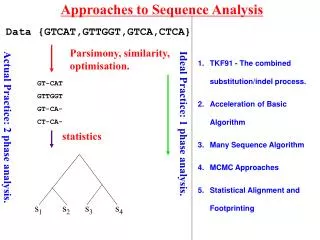
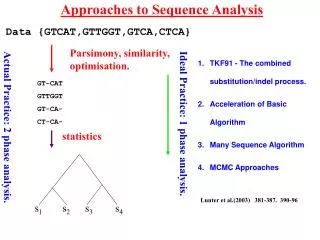
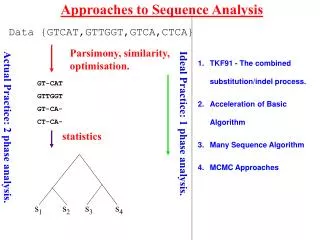
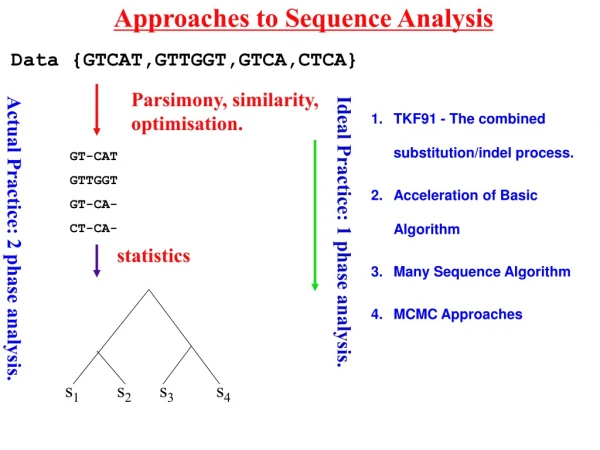
Evolutionary Analysis • Inferring phylogenies – finding the tree that implies the correct evolutionary history of the sequences • Principal Methods: • Parsimony analysis • Distance methods • Maximum Likelihood • Each approach has its own strengths/weaknesses • To assess the “correctness” of the tree we need to understand: • Overall signal & noise in the data • How the tree comparisons to alternate trees • How reliable the individual branches are These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Evolutionary Analysis • Refining Trees • Bootstrap analysis – can give us estimates of variability • Incorporating information about duplication and loss: • reconcile a gene tree with a species tree; • identify gene duplications • root an unrooted tree by minimizing gene duplications and losses; • refine rooted trees to minimize duplications and losses • Groups analysis • Discover what is unique about each subgroup in a tree • Help resolve which subgroup a sequence belongs to in a tree These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns Project Homology Modeling CURATED DATASET These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Workshop Project – Part I • Select a sequence family that you are familiar with • Select the Estradiol 17 beta Dehydrogenase superfamily • The PIR Superfamily is PIRSF000095 • Select the Haloalkane Dehalogenase superfamily • The PIR Superfamily is PIRSF037173 These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Workshop Project – Part II • Collect an initial set of sequences: • Perform a database search with the query sequence across several databases with different algorithms and different parameters • Use iProClass to collect an initial set of related sequences. • Generate a multiple sequence alignment • Perform a multiple sequence alignment with a variety of different alignment algorithms • Select “best” multiple sequence alignment These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Workshop Project – Part III • Improve the quality of your alignment: • Search for local patterns in the group of sequences • Refine multiple sequence alignment based on local patterns • Identify additional family members: • Convert alignment into HMM and a PSSM • Search the database for distantly related sequences. • What is the most distantly related sequence you can find? These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center
Workshop Project – Part IV • Add evolutionary information: • Use your MSA to help build an initial phylogenetic tree • Refine the phylogenetic tree using bootstrapping • Compare the gene tree and species tree • Identify key residues that define subgroups groups of sequences • Add structural information: • Visualize the key residues/motifs on a representative structure • Produce and visualize a structure using homology modeling techniques These materials were developed with funding from the US National Institutes of Health grant #2T36 GM008789 to the Pittsburgh Supercomputing Center