Download
1 / 16
160 likes | 262 Views
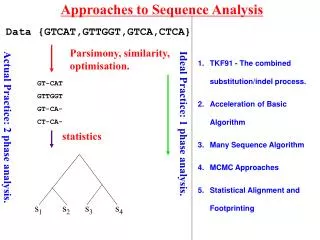
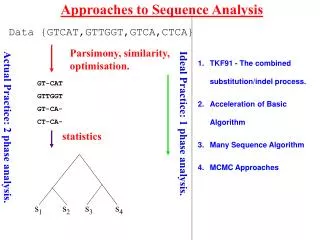
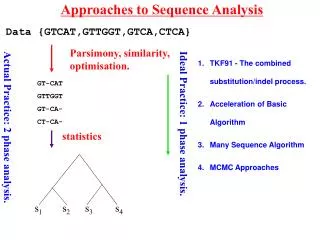
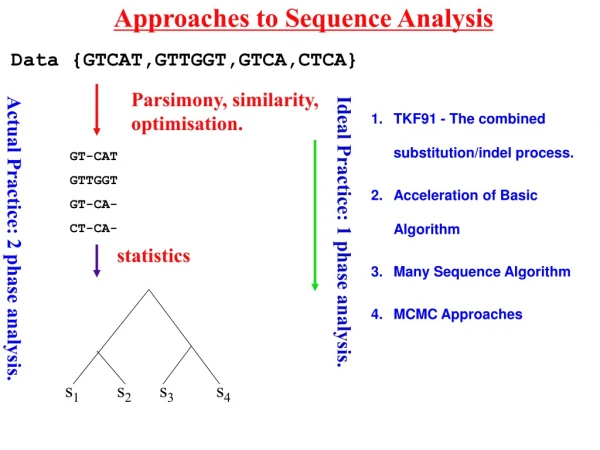
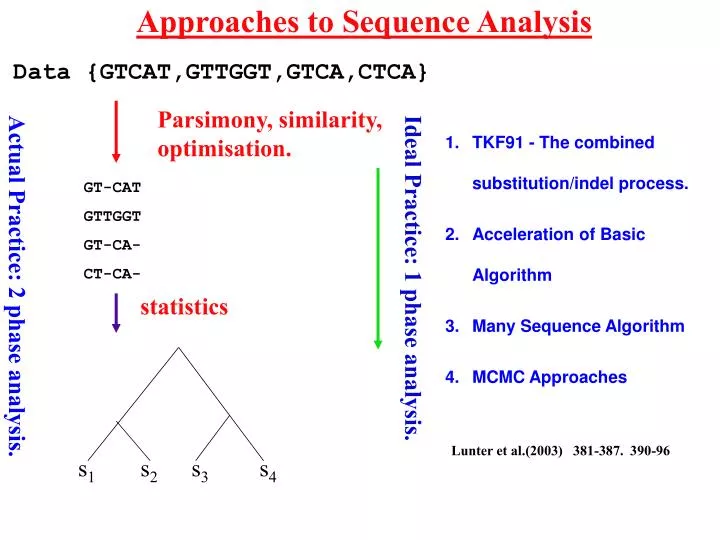
Approaches to Sequence Analysis. Data {GTCAT,GTTGGT,GTCA,CTCA}. Parsimony, similarity, optimisation. TKF91 - The combined substitution/indel process. Acceleration of Basic Algorithm Many Sequence Algorithm MCMC Approaches. GT-CAT GTTGGT GT-CA- CT-CA-. Ideal Practice: 1 phase analysis.

E N D
Approaches to Sequence Analysis Data {GTCAT,GTTGGT,GTCA,CTCA} Parsimony, similarity, optimisation. TKF91 - The combined substitution/indel process. Acceleration of Basic Algorithm Many Sequence Algorithm MCMC Approaches GT-CAT GTTGGT GT-CA- CT-CA- Ideal Practice: 1 phase analysis. Actual Practice: 2 phase analysis. statistics Lunter et al.(2003) 381-387. 390-96 s1 s2 s3 s4
T= 0 # - - - ## # # # T = t # # # # s1 r s2 s1 s2 s1 s2 Thorne-Kishino-Felsenstein (1991) Process A # C G * • (birth rate) < m(death rate) 1. P(s) = (1-l/m)(l/m)l pA#A* .. *pT #T l =length(s) 2. Time reversible:
# - - - - - # # # # k * - - - - * # # # # k l & m into Alignment Blocks A. Amino Acids Ignored: # - - - ## # # k e-mt[1-lb](lb)k-1 [1-lb-mb](lb)k [1-lb](lb)k p’k(t) pk(t) p’’k(t) b=[1-e(l-m)t]/[m-le(l-m)t] p’0(t)= mb(t) B. Amino Acids Considered: T - - - RQ S W Pt(T-->R)*pQ*..*pW*p4(t) 4 • T - - - - • R Q S WpR *pQ*..*pW*p’4(t) • 4
Dpk = Dt*[l*(k-1) pk-1 + m*k*pk+1 - (l+m)*k*pk] Dp’k=Dt*[l*(k-1) p’k-1+m*(k+1)*p’k+1-(l+m)*k*p’k+m*pk+1] Dp’’k=Dt*[l*k*p’’k-1+m*(k+1)*p’’k+1- [(k+1)l+km]*p’’k] Differential Equations for p-functions # - - ... - # # # ... # # - - - ... - - # # # ... # * - - - ... - * # # # ... # Initial Conditions: pk(0)= pk’’(0)= p’k (0)= 0 k>1 p1(0)= p0’’(0)= 1. p’0 (0)= 0
Basic Pairwise Recursion (O(length3)) i j Survives: Dies: i-1 i i-1 i j-1 j j i-1 i i j-2 j i-1 j j-1 …………………… …………………… …………………… e-mt[1-lb](lb)k-1, where …………………… …………………… b=[1-e(l-m)t]/[m-le(l-m)t] 0… j (j+1) cases 1… j (j) cases
Basic Pairwise Recursion (O(length3)) survive death j (i-1,j) j-1 (i-1,j-1) Initial condition: p’’=s2[1:j] ………….. (i-1,j-k) ………….. ………….. i-1 i (i,j)
Corner Cutting ~100-1000 Better Numerical Search ~10-100 Ex.: good start guess, 28 evaluations, 3 iterations Accelleration of Pairwise Algorithm (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) Simpler Recursion ~3-10 Faster Computers ~250 1991-->2000 ~106
a-globin (141) and b-globin (146) (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) 430.108 : -log(a-globin) 327.320 : -log(a-globin -->b-globin) 747.428 : -log(a-globin, b-globin) = -log(l(sumalign)) l*t: 0.0371805 +/- 0.0135899 m*t: 0.0374396 +/- 0.0136846 s*t: 0.91701 +/- 0.119556 E(Length) E(Insertions,Deletions) E(Substitutions) 143.499 5.37255 131.59 Maximum contributing alignment: V-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS--H---GSAQVKGHGKKVADALT VHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFS NAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR DGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH Ratio l(maxalign)/l(sumalign) = 0.00565064
VLSPADNAL.....DLHAHKR 141 AA long *########### …. ### 141 AA long 2 108 years 2 107 years 2 109 years *########### …. ### *########### …. ### ???????????????????? k AA long 109 years The invasion of the immortal link
Algorithm for alignment on star tree (O(length6))(Steel & Hein, 2001) *ACGC *TT GT s2 s1 a s3 *ACG GT *###### * (l/m)
Binary Tree Problem a1a2 * * # # # - - # # # - # TGA ACCT s1 s3 a1 a2 s2 s4 GTT ACG • The ancestral sequences & their alignment was known. ii. The alignment of ancestral alignment columns to leaf sequences was known The problem would be simpler if: How to sum over all possible ancestral sequences and their alignments?: A Markov chain generating ancestral alignments can solve the problem!!
- # # E # # - E * * lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # # lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) _ #lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # - lb Generating Ancestral Alignments a1 * a2 * # # l/m (1- lb)e-m E E (1- l/m) (1- lb) - # lb
The Basic Recursion ”Remove 1st step” - recursion: S E ”Remove last step” - recursion: Last/First step removal are inequivalent, but have the same complexities. First step algorithm is the simplest.
Sequence Recursion: First Step Removal Pa(Sk): Epifixes (S[k+1:l]) starting in given MC starts in a. Pa(Sk) = Where P’(kS i,H) = F(kSi,H)
Maximum likelihood phylogeny and alignment Gerton Lunter Istvan Miklos Alexei Drummond Yun Song Human alpha hemoglobin;Human beta hemoglobin; Human myoglobin Bean leghemoglobin Probability of data e-1560.138 Probability of data and alignment e-1593.223 Probability of alignment given data 4.279 * 10-15 = e-33.085 Ratio of insertion-deletions to substitutions: 0.0334
Metropolis-Hastings Statistical Alignment Lunter, Drummond, Miklos, Jensen & Hein, 2005