Download

1 / 28
280 likes | 383 Views
Sekvensklassificering & Hidden Markov Models. Bioinformatik 3p 24 Januari 2002 Patrik Johansson, Inst. f ör Cell & Molekylärbiologi, Uppsala Universitet. Craig. ?. Klassificering baserat p å deterministiska regler. Konsensussekvenser (kvalitativ / fix längd)

E N D
Sekvensklassificering & Hidden Markov Models Bioinformatik 3p 24 Januari 2002 Patrik Johansson, Inst. för Cell & Molekylärbiologi, Uppsala Universitet
Craig ?
Klassificering baserat på deterministiska regler • Konsensussekvenser (kvalitativ / fix längd) • AT{LV}G{CA} ATLGC, ATLGA, ATVGC, ATVGA • Weight matrices (kvantitativ / fix längd) • ex. GYCG, GYGG, GYCC
Klassificering baserat på deterministiska regler.. • Prosites (kvalitativ / variabel längd) • G{YF}W(3,5)NH GYWACLNH, GFWSFAEVNH ... • Profiles (kvantitativ / variabel längd) • Positionsspecifik viktmatris + Gap penalty
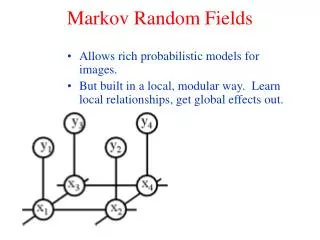
A A A B B B C C C Hidden Markov Models, introduktion • Generell metod för mönsterigenkänning, jmfr. Profiles & neurala nätverk • En Markovkjedja av händelser • Tre mynt A, B & C CAABA.. Sekvens, ex. Heads Heads Tails, genereras av gömd Markovkjedja
A A B A B B C C C Heads Tails Tails Hidden Markov Models, introduktion.. • En HMM genererar sekvenser / sekvensdistributioner • Emitterar symbol (T, H) i varje state baserat på en emissionssannolikhet ei Väljer nästföljande state utifrån • någon övergångssannolikhet ai,j • Ex. sekvensen ‘Tails Heads Tails’
M1 Mj MN B E Profile Hidden Markov Model arkitektur • En första approach för • sekvensdistributionsmodellering
Mj - Mj Mj+ Ij B E Profile Hidden Markov Model arkitektur.. • Modellering av insertioner ejI(a) =q(a) , där q(a) är bakgrundsdistributionen av aminosyror
Mj Mj Dj Profile Hidden Markov Model arkitektur.. • Modellering av deletioner
Dj Ij E B Mj Profile Hidden Markov Model arkitektur.. Insert & deletestates generaliseras till alla positioner. Modellen genererar sekvenser från state Bgenom succesiva emissioner och transitioner tills state E nås
Probabilistisk sekvensmodellering • Klassificeringskriterium ( 1 ) Bayes sats ; ( 2 ) ..men, P(M) & P(s)..? ( 3 )
Probabilistisk sekvensmodellering.. Om N tillåts modellera hela sekvensrymden tex. genom N = q ; ( 4 ) Eftersom , logaritmsannolikheter smidigare Def., log-odds score ; ( 5 )
Probabilistisk sekvensmodellering.. Ekv. ( 4 ) & ( 5 ) ger ett nytt klassificeringskriterium ; ( 6 ) Milosavljevics algoritmsignifikanstest ..för viss signifikansnivå , dvs. antalet felaktigt klassificerade sekvenser av en n stor databas, krävs visst threshold d ( 7 )
Probabilistisk sekvensmodellering.. Exempel. Om z=e eller z=2, signifikansnivån väljs till en felaktig klassificering, en false positive, per tiotusen och vi klassificerar n=1000 sekvenser ; bits nits, Man brukar definiera känslighet, ‘hur många hittas’ ; ..och selektivitet, ‘hur många är korrekta’ ;
Modellkonstruktion • Från initial alignment • Vanligaste metoden, utgå från multipel alignment av tex. en proteinfamilj • Iterativt • Genom att sucessivt söka i en databas och inkorporera nya sekvenser över något visst threshold till modellen • Neuralt inspirerad • Modellen tränas mha. någon kontinuerlig minimerings- algoritm, ex. Baum-Welsh eller Steepest Descent
D2 I2 M3 B M2 M1 D2 I2 D2 M3 E E E E B M2 M1 I2 M3 B M2 M1 D2 I2 M3 B M2 M1 Modellkonstruktion.. Initial alignment, potentiella matchstates markerade med ()
Dj-1 Ij-1 Mj-1 Mj Evaluering av sekvenser Den optimala alignmenten, dvs. den path som har störst sannolikhet att generera sekvensen s, beräknas mha. dynamisk programmering Det maximala log-odds scoret VjM(si) för matchstate j som emitterar aminosyra si beräknas från emissionsscore, föregående maxscore och transitionsscore
Evaluering av sekvenser.. Viterbis Algoritm, ( 8 ) ( 9 ) ( 10 )
Parameterestimering, bakgrund Proteiner med liknande struktur och funktion kan ha mycket olika sekvenser Klassisk sekvensalignment baserad på heuristiska parametrar klarar inte en sekvensidentitet under ~ 50-60% Substitutionsmatriser för in statisk a priori information om aminosyror och proteinsekvenser korrekta alignments ned till ~ 35% sekvensidentitet, ex. CLUSTAL Hur komma längre ned I ‘the twilight zone’..? - Mer och dynamisk a priori information..!
Parameterestimering Vad är sannolikheten att emittera alanin I första matchstatet, eM1(‘A’)..? • Maximum likelihood-estimering
Parameterestimering.. • Add-one pseudocount estimering • Background pseudocount estimering
Parameterestimering.. • Substitutionmixture estimering • Score : Maximum likelihood ger pseudocounts : Total estimering :
Parameterestimering.. • Ovanstående metoder är trots sin dynamiska utformning till viss del baserade på heuristiska parametrar. • Metod som kompenserar och kompletterar eventuell brist på data på ett statistiskt korrekt sätt ; • Dirichlet mixture estimering Bayes sats beskriver hur a priori information A värderas I vissa strukturella omgivningar verkar det finnas några vanligt förekomande aminosyradistributioner inte bara en, bakgrundsdistributionen q Antar att det finns k st probabilitetsdensiteter som genererar dessa
Parameterestimering, Dirichlet Mixtures En metod som gör en separat estimering för alla j=1..k komponenter och sedan viktar ihop dem med sannolikheten att vår countvektor är genererad just av den komponenten Om de k komponenterna modelleras från en kurerad databas med alignments tex. som sk. Dirichlet densiteter kan ett explicit uttryck för sannolikheten att genererats från komponent j ställas upp mha. Bayes sats
Parameterestimering, Dirichlet Mixtures.. De k komponenterna ger upphov till toppar av aa-distributioner i någon multidimensionell sekvensrymd Beroende på var i sekvensrymden vår countvektor n ligger, dvs beroende på vilka komponenter som antas har genereratn, inkorporeras distributionsinformation från dessa
Klassifikationsexempel Alignment av några kända Glykosidhydrolassekvenser från familj GH16 • Definiera vilka kolumner som ska utgöra basis för matchstates • Ställ upp den korresponderande HMM-grafen för vår modell M • Estimera emissions- och transitionssannolikheter, ej& ajk • Evaluera sannolikheten / log-odds scoret att en viss sekvens s från någon databas har genererats av M med hjälp av Viterbis algoritm • Ifall score(s | M) > d, kan sekvensen s klassificeras till familjen GH16
Klassifikationsexempel.. En viss sekvens s1=WHKLRQevalueras och får ett score på -17.63 nits, dvs. sannolikheten att modellen M har genererat s1 är mycket liten En annan sekvens s2=SDGSYT får ett score på 27.49 nits och kan med god signifikans klassificeras till familjen
Sammanfattning • Hidden Markov Models används dels för klassificering / sökning (PFAM) och dels för sekvensmappning / homologimodellering • Till skillnad från vanlig alignment används en positionsspecifik approach för sekvens-, insertions- och deletionsdistributioner • Desto mer a priori information som inkorporeras, desto större känslighet men mindre selektivitet. Analogt för omvändningen