Download

1 / 76
820 likes | 1.43k Views
Inflammatory Myopathies. Susan Wallis, MD. Idiopathic inflammatory myopathies. Polymyositis Dermatomyositis Juvenile dermatomyositis Inclusion body myositis Myositis associated with collagen vascular disease Myositis associated with malignancy. Idiopathic inflammatory myopathies.

E N D
Inflammatory Myopathies Susan Wallis, MD
Idiopathic inflammatory myopathies • Polymyositis • Dermatomyositis • Juvenile dermatomyositis • Inclusion body myositis • Myositis associated with collagen vascular disease • Myositis associated with malignancy
Idiopathic inflammatory myopathies • Polymyositis • Dermatomyositis • Juvenile dermatomyositis • Inclusion body myositis • Myositis associated with collagen vascular disease • Myositis associated with malignancy
Inflammatory myopathies • Rare heterogeneous group of acquired diseases characterized by inflammatory infiltrate of skeletal muscle. • Incidence of about 2-10 per 1 million people per year in the United States. • Potentially treatable.
Polymyositis/Dermatomyositis • Heliotrope rash was first described in 1875 in France. • In 1888 the first American biopsy documented polymyositis in ruling out Trichinella. • 1930 Gottron reported skin lesions • 1967 the pathology of inclusion body myositis was described. Hochberg et al. Rheumatology 3rd ed. 2003
Epidemiology • Bimodal age distribution in PM/DM • Between 10-15 years in children • Between 45-60 in adults • Inclusion body • More common after age 50 years • Female predominance
Drugs and toxins: Chloroquine Colchicine Corticosteroids Heroin Alcohol Fibrates/statins AZT Metabolic Malignancy Genetic HLA-DRB1 HLA-DQA1 TNF2(-308) Infectious agents: Bacteria Staphylococci Clostridia Rickettsias Mycobacteria Parasites Toxoplasma Trichnella Schistosoma Cysticerca Borrelia Viruses Coxsackie Echo Influenze Adeno Differential diagnosis
Criteria to define polymyositis and dermatomyositis proposed by Bohan and Peter • Symmetric weakness of limb girdle muscles and anterior neck flexors. 2. Skeletal muscle histologic examination showing evidence of necrosis of types I and II muscle fibers, phagocytosis, regeneration with basophilia, large sarcolemmal nuclei and prominent nucleoli, atrophy in a perifascicular distribution, variation in fiber size, and an inflammatory exudate. N Engl J Med 292:344, 1975
Elevation of levels of serum skeletal muscle enzymes • Electromyographic (EMG) triad of short, small polyphasic motor units; fibrillations, positive waves, and insertional irritability; and bizarre high-frequency discharges. 5. Dermatologic features including a heliotrope rash with periorbital edema; a scaly, erythematous dermatitis over the dorsa of the hands, especially over the MCP and PIP joints (Gottron's sign); and involvement of the knees, elbows, medial malleoli, face, neck, and upper torso.
Pathologic criteria Electron microscopy: Microtubular filaments in the inclusions. Light microscopy: Lined vacuoles Intranuclear or intracytoplasmic inclusions or both Clinical criteria Proximal muscle weakness Distal weakness EMG evidence of generalized myopathy Increase in serum muscle enzymes Failure of muscle weakness to improve on high-dose steroids Diagnostic criteria for IBM
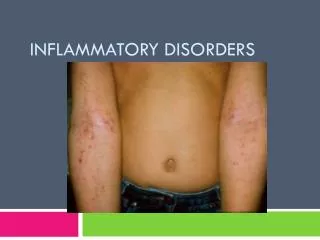
Polymyositis/Dermatomyositis • Occur sporadically or in association with other systemic autoimmune disease • More common in women than men. • DM common than PM. • DM can clinically manifest with heliotrope rash, Grotton’s papules, shawl rash, erythematous nailfolds, dermatomyositis sine myositis.
Clinical features • Progressive painless weakness • Difficulty lifting above head/combing hair • Difficulty arising from a low chair or toilet • Nasal regurgitation or choking when eating • Hoarseness, change in voice • *Ocular/facial muscle involvement is very uncommon • Fatigue • Fever
Other clinical features • Weight loss • Nonerosive inflammatory polyarthritis in rheumatoid-like distribution • Except in Jo-1 positive, can be erosive and deforming. • Raynaud’s phenomenon • Interstitial lung disease • Cardiac abnormalities • Amyopathic dermatomyositis
Inclusion body myositis • Can present with features identical to PM. • Onset is typically insidious and progression is slow. • May differ from PM in that it may include focal, distal or asymmetric weakness. • Dyspagia is a late occurrence. • CK only slightly increased and can be normal in up to 25% of patients.
Dermatologic manifestations www.jfponline.com/Pages.asp?AID=2763&UID=
Nailfold capillaries www.hakeem-sy.com/main/files/images/20_2.jpg
Cardiac • Myocarditis • With secondary arrhythmias and CHF • Myocardial fibrosis • Cor pulmonale • Secondary to ILD • Accelerated atherosclerosis associated with prolonged steroid use
Dyspnea • Non-pulmonary: respiratory muscle weakness, cardiac involvement • Pulmonary: • ILD: NSIP, UIP, diffuse alveolar damage, cryptogenic organizing pneumonia • Pulmonary hypertension • Alveolar hemorrhage • Infection: with or without aspiration • Drug induced
Pulmonary evaluation • CT scan • Increased interstitial markings • PFTs • Decreased TLV and DLCO • BAL • Abnormal number of leukocytes • Biopsy • Mononuclear cell infiltration, destruction of alveolar spaces and fibrosis
GI Tract • Pharyngeal muscle involvement • Dyphonia • Dysphagia • Postprandial symptoms of bloating, pain and distension • Pneumatosis cystoides intestinalis
Malignancy risk • Strong association between malignancy and dermatomyositis, but less clearly with polymyositis. • Ovarian, lung, pancreatic, stomach and colorectal and non-Hodgkin lymphoma • The overall risk is greatest in the first 3 years after diagnosis but is still increased through all years of follow-up.
Inflammation • Dermatomyositis • B cells and CD4 are abundant in the pervascular region. • MAC found in the perivascular areas and within intrafascicular capillaries • Damage to intrafascicular capillaries • Polymyositis and inclusion body myositis • Normal appearing muscle cells are invaded by T cells • PM/DM • Increased expression of costimulatory molecules
Polymyositis pleiad.umdnj.edu/.../muschtml/musc008.htm Endomysial inflammatory infiltrate surrounding and invading non-necrotic muscle fibers www.neuropathologyweb.org/chapter13/chapter13
Dermatomyositis Necrotic and regenerating muscle fibers in perifascicular regions www.neuro.wustl.edu/.../pathol/dermmyo.htm www.phoenixneurology.com
Pathogenesis • Humoral • Autoantibodies • Directed against cell components • Directed at intracellular, ususally intracytoplasmic molecules • Usually part of the protein synthesis machinery • Cellular • Genetic
Autoantibodies • Autoantibodies have been identified in patients with myositis. • Not seen in inclusion body myositis • Can help predict specific syndromes. • Differentiate between types of idiopathic myositis versus myositis associated with other conditions.
Autoantibodies • Myositis specific antibodies (MSA) • Present in 30-60% of patients with PM/DM • Anti-aminoacyl-tRNA synthetases (ARS). • Anti-SRP • Anti-Mi-2 Autoimmunity, 2006;39(3):161-170
Antisynthetase syndrome • Aminoacyl-tRNA-synthetase is a cytoplasmic enzyme involved in aminoacylation. • The most common ARS is histidyl-RNA-synthetase, also called Jo-1. www.arodia.com/.../orderByAttribute__caption
Common characteristics • Myopathy • Interstitial lung disease • Raynaud’s phenomenon • Polyarthritis • Fever • Mechanic’s hands
Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies • Aim of the study to determine if these antibodies were predictive of clinical course of ILD in idiopathic inflammatory myositis patients. • Retrospective study of 74 patients who met Peter-Bohan criteria. • The patients with ILD have a worse prognosis than those without. • Anti-ARS are strongly associated with ILD Autoimmunity 2006; 39(3):233-241
Prevalence of symptoms of patients with antisynthetase syndrome Autoimmunity 2006; 39(3):233-241
Interstitial lung disease Autoimmunity 2006; 39(3): 233-241
Anti-SRP Antibodies • Cytoplasmic antibody • SRP is an RNA-protein complex that binds newly synthesized proteins and guides them to the endoplasmic reticulum for translocation.
Clinical • Very rare • Chiefly proximal muscle involvement with rhabdomyolysis • Usually poor response to steroids • ILD possible but uncommon • Skin and joints spared Joint, Bone, Spine. 2006;73:646-654
Anti-Mi-2 • Antibodies directed to a nuclear macromolecular complex involved in transcription. • Strong specificity for dermatomyositis. • Usually good response to treatment.
Myositis Associated Antibodies • Anti-PM-Scl • Anti-RNP • Anti-Ro • Anti-La • Anti-Ku
Myositis-associated antigens Autoimmunity, May 2006; 39(3): 161–170
Anti-PM-Scl antibodies • Directed against a nucleolar macromolecular complex • Primarily polymyositis or dermatomyositis/scleroderma overlap • Strongly associated with HLA-DR3 • Seen in 5-25% of patients with myositis.