Download

1 / 32
320 likes | 448 Views
Learn about collagen and elastin, key fibrous proteins in the body, with unique mechanical properties due to their amino acid structure. Discover how collagen supports tissues like skin, tendons, and blood vessels, and the different types of collagens found in the human body.

E N D
I. OVERVIEW • Collagenand elastinare examples of common, well-characterized fibrous proteins that serve structural functions in the body. • For example, collagenand elastinare found as components of skin, connectivetissue, bloodvesselwalls, and sclera and cornea of the eye.
I. OVERVIEW • Each fibrousproteinexhibits special mechanical properties, resulting from its unique structure, which are obtained by combining specific amino acids into regular, secondary structural elements. • This is in contrast toglobularproteins, whose shapes are the result of complex interactions between secondary, tertiary, and sometimes, quaternary structural elements.
II. Collagen • Collagenis the most abundant protein in the human body. • A typical collagen molecule is a long, rigidstructurein which threepolypeptides "-chains" are wound around one another in a rope-like triple-helix.
II. Collagen • Although collagenmolecules are found throughout the body, their types and organization are dictated by the structural role collagenplays in a particular organ. • In some tissues, collagenmay be dispersed as a gel support to the structure, as in the extracellular matrixor the vitreous humor of the eye. • In other tissues, collagenmay be bundled in tight, parallel fibers great strength, as in tendons. • In the cornea of the eye, collagenis stacked transmit light with a minimum of scattering. • Collagen of bone occurs as fibers arranged at an angle to each other resist mechanical shear from any direction.
A. Types of collagen • The collagensuperfamily of proteins includes more than 20collagen types, and proteins that have collagen-like domains. • The three polypeptide -chains are held together by hydrogenbondsbetween the chains. • Variations in the amino acid sequence of the -chains structural components that are about the same size (approximately 1000 amino acids long), but with slightly different properties. • These -chains are combined to form the various types of collagen found in the tissues.
A. Types of collagen • The most common collagen: • Type I, contains twochains called l and onechain called 2 (122) • Type II collagen contains threel chains (13). • The collagens can be organized into threegroups, based on their location and functions in the body.
1. Fibril-forming collagens: • Types I, II, and III are the fibrillarcollagens, and have the rope-like structure. • In the electron microscope, these linear polymers of fibrilshave characteristic banding patterns reflecting the regular staggered packing of the individual collagen molecules in the fibril.
1. Fibril-forming collagens • Type I collagenfibers are found in supporting elements of high tensile strength (e.g. tendon and cornea). • Fibers formed from type II collagen molecules are restricted to cartilaginous structures. • Fibrilsderived from type III collagenare prevalent in more distensible tissues, such as blood vessels.
2. Network-forming collagens • Types IV and VII form a three-dimensional mesh, rather than distinct fibrils. • For example, type IV molecules assemble into a sheet or meshwork that constitutes a major part of basement membranes. • [ Basement membranes are thin, sheet-like structures that provide mechanical support for adjacent cells, and function as a semipermeable filtration barrier for macromolecules in organs such as the kidney and the lung.]
3. Fibril-associated collagens: • Types IX and XII bind to the surface of collagenfibrils, linking these fibrilsto one another and to other components in the extracellular matrix.
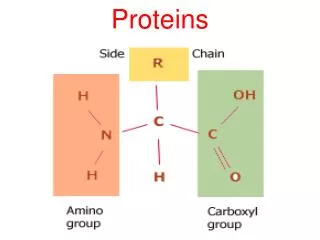
B. Structure of collagen • Amino acid sequence: • Collagen is rich in prolineand glycine, both of which are important in the formation of the triple-stranded helix. • Proline facilitates the formation of the helical conformation of each -chain because its ring structure "kinks" in the peptide chain. • Glycine, the smallest amino acid, is found in every thirdposition of the polypeptide chain. • It fits into the restricted spaces where the three chains of the helix come together.
B. Structure of collagen • The glycineresidues are part of a repeating sequence. —Gly—X—Y—, where X is frequently prolineand Y is often hydroxyprolineor hydroxylysine. • Most of the .- chain can be regarded as a polytripeptide whose sequence can be represented as (—Gly—Pro—Hyp—)
2. Triple-helical structure: B. Structure of collagen • Unlikemostglobular proteins that are folded into compact structures, collagen, a fibrousprotein, has an elongated, triple-helical structure that places many of its amino acid side chains on the surface of the triple-helical molecule. • [This allows bond formation between the exposed R-groups of neighboring collagen monomers aggregation into long fibers]
3. Hydroxyproline and hydroxylysine: B. Structure of collagen • Collagencontains hydroxyproline(hyp) and hydroxylysine (hyl), which are not present in most other proteins. • These residues result from the hydroxylation of some of the prolineand lysineresidues after their incorporation into polypeptide chains. • The hydroxylation is, thus, an example of posttranslational modification . • Hydroxyprolineis important in stabilizing the triple -helical structure of collagenbecause it maximizes interchain hydrogen bond formation.
4. Glycosylation: B. Structure of collagen • The hydroxyl group of the hydroxylysineresidues of collagenmay be enzymatically glycosylated. • Most commonly, glucose and galactose are sequentially attached to the polypeptide chain prior to triple-helix formation.
C. Biosynthesis of collagen • The polypeptide precursors of the collagenmolecule are formed in fibroblasts(or in the related osteoblastsof bone and chondroblastsof cartilage), and are secreted into the extracellular matrix. • After enzymic modification, the mature collagenmonomers aggregate and become cross-linked collagen fibrils.
D. Degradation of collagen • Normal collagensare highly stable molecules, having half-lives as long as several months. • However, connective tissue is dynamic and is constantly being remodeled, often in response to growth or injury of the tissue. • Breakdown of collagenfibrils is dependent on the proteolyticaction of collagenases, which are part of a large family of matrix metalloproteinases. • Type I collagen: the cleavage site is specific, generating three-quarter and one-quarter length fragments. • These fragments are further degraded by other matrix proteinases to their constituent amino acids.
E. Collagen diseases • Defects in any one of the many steps in collagen fiber synthesis genetic disease inability of collagen to form fibers properly and provide tissues with the needed tensile strength normally provided by collagen. • > 1000 mutations have been identified in 22 genes coding for 12 of the collagen types. • The following are examples of diseases that are the result of defective collagen synthesis.
Ehlers-Danlos syndrome (EDS) • This disorder is a heterogeneous group of generalized connective tissue disorders that result from inheritable defects in the metabolism of fibrillarcollagenmolecules. • EDS can result from a deficiency of collagen-processing enzymes(for example, lysyl hydroxylase or procollagenpeptidase), or from mutations in the amino acid sequences of collagentypes I, III, or V. • The most clinically important mutations are found in the gene for type III collagen. Collagencontaining mutant chains is not secreted, and is either degraded or accumulated to high levels in intracellular compartments. • Because collagen type III is an important component of the arteries, potentially lethal vascular problems occur. [Note: Although collagen type III is only a minor component of the collagen fibrils in the skin, patients with EDS also show, for unknown reasons, defects in collagen type I fibrils. This results in fragile, stretchy skin and loose joints.]
Osteogenesis imperfecta (OI) • This disease, known as brittle bone syndrome, is also a heterogeneous group of inherited disorders distinguished by bones that easily bend and fracture. • Retarded wound healing and a rotated and twisted spine leading to a “humped-back” (kyphotic) appearance are common features of the disease. • Type I OI is called osteogenesisimperfectatarda. • The disease is the consequence of decreased production of α1 and α2 chains. It presents in early infancy with fractures secondary to minor trauma, and may be suspected if prenatal ultrasound detects bowing or fractures of long bones.
Osteogenesis imperfecta (OI) • TypeIIOI is called osteogenesisimperfectacongenita, and is the most severe. • Patients die of pulmonary hypoplasia in utero or during the neonatal period. • Most patients with severe OI have mutations in the gene for either the pro-α1 or pro-α2 chains of type I collagen. • The most common mutations cause the replacement of glycineresidues (in –Gly–X–Y–) by amino acids with bulky side chains. • The resultant structurally abnormal pro-αchains prevent the formation of the required triple-helical conformation.
ELASTIN • In contrast to collagen, which forms fibers that are tough and have high tensile strength, Elastinis a connective tissue protein with rubber-like properties. • Elastic fibers, composed of elastinand glycoproteinmicrofibrils, are found in the lungs, the walls of large arteries, and elastic ligaments. • They can be stretched to several times their normal length, but recoil to their original shape when the stretching force is relaxed.
A. Structure of elastin • Elastinis • an insoluble protein polymer • synthesized from a precursor, tropoelastin, ( a linear polypeptide composed of about 700 amino acids that are primarily small and nonpolar e.g. glycine, alanine, and valine). • Elastinis also, • rich in prolineand lysine. • contains a little hydroxyproline. • contains NOhydroxylysine.
A. Structure of elastin • Tropoelastinis secreted by the cell into the extracellular space. • There, it interacts with specific glycoprotein microfibrils, such as fibrillin, which function as a scaffold onto which tropoelastinis deposited. • Mutations in the fibrillin gene are responsible for Marfan’s syndrome.
A. Structure of elastin • Some of the lysylside chains of the tropoelastinpolypeptides are oxidativelydeaminated by lysyl oxidase forming allysine residues. • 3 of the allysyl side chains + one unaltered lysyl side chain from the same or neighboring polypeptides form a desmosine cross-link. • This produces Elastin- an extensively interconnected, rubbery network that can stretch and bend in any direction when stressed connective tissue elasticity.
B. Role of 1-antitrypsin in elastin degradation • 1-antitrypsin: • Blood and other body fluids contain a protein, 1- antitrypsin (1-AT) or ( 1-antiproteinase) inhibits a number of proteolytic enzymes (proteasesor proteinases) hydrolyze and destroy proteins. • [The inhibitor was originally named 1-antitrypsinbecause it inhibits the activity of trypsin (a proteolytic enzyme synthesized as trypsinogen by the pancreas] • 1- AT comprises > 90% of the 1-globulin fraction of normal plasma.
1-antitrypsin: • 1- AT has the important physiologic role of inhibiting neutrophil elastase- a powerful proteasethat is released into the extracellular space, and degrades elastinof alveolar walls, as well as other structural proteins in a variety of tissues. • Most of the 1-AT found in plasma is synthesized and secreted by the liver. • The remainder is synthesized by several tissues, including monocytes and alveolarmacrophages, which may be important in the prevention of local tissue injury by elastase.
2. Role of 1-AT in the lungs: • In the normal lung, the alveoli are chronically exposed to low levels of neutrophil elastasereleased from activated and degenerating neutrophils. • This proteolytic activity can destroy the elastinin alveolar walls if unopposed by the inhibitory action of 1-AT( the most important inhibitor of neutrophil elastase). • Because lung tissue cannotregenerate, emphysema results from the destruction of the connective tissue of alveolar walls.
3. Emphysema resulting from 1-AT deficiency: • In USA, inherited defects in 1-AT 2-5% of patients having emphysema. • A number of different mutations in the 1-AT gene deficiency of this protein, but one single purine base mutation (GAG AAG, substitution of lysine for glutamic acid at position 342 of the protein) is clinically the most widespread.
3. Emphysema resulting from 1-AT deficiency: • An individual must inherit 2 abnormal 1- AT alleles to be at risk for the development of emphysema. • In a heterozygote (with one normal and one defective gene) the levels of 1-AT are sufficient to protect the alveoli from damage. • A specific 1-AT methionine is required for the binding of the inhibitor to its target proteases. • Smoking oxidation and subsequent inactivation of that methionine residue, render the inhibitor powerless to neutralize elastase. • Smokers with 1 -AT deficiency, have rate of lung destruction poorer survival rate than nonsmokers with the deficiency.
Summary • Collagenand elastinare fibrous proteins. • Collagenmolecules contain an abundance of proline, lysine, and glycine, the latter occurring at every third position in the primary structure. • Collagenalso contains hydroxyproline, hydroxylysine, and glycosylated hydroxylysine, each formed by posttranslational modification. • Collagenmolecules typically form fibrils containing a long, stiff, triple-stranded helical structure, in which three collagenpolypeptide chains are wound around one another in a rope-like superhelix (triple helix). • Other types of collagen form mesh-like networks. • Elastinis a connective tissue protein with rubber-like properties in tissues such as the lung. • α1-Antitrypsin(α1-AT), produced primarily by the liver but also by tissues such as monocytes and alveolarmacrophages, prevents elastindegradation in the alveolar walls. A deficiency of α1-AT can cause Emphysema and, in some cases, cirrhosis of the liver.